
Abstract
Endogenous glycosaminoglycans (GAGs) with a similar structure to heparin are widely distributed in various tissues. A fluorescence probe, namely Heparin Red, can detect polyanionic GAGs in plasma samples. The purpose of this study is to measure endogenous GAGs in various plasma samples obtained from different pathologic states in comparison to healthy controls utilizing this method. Plasma samples were obtained from patient groups including atrial fibrillation (AF), end-stage-renal-disease (ESRD), diabetes mellitus (DM), sepsis, cancer, liver disease (LD), and pulmonary embolism (PE). Normal human plasma (NHP) was used as healthy controls. The Heparin Red kit from Red Probes (Münster, Germany) was used for the quantification of endogenous GAGs in each sample before and after heparinase I degradation. All results were compiled as group means ± SD for comparison. NHP was found to have relatively low levels of endogenous GAGs with a mean concentration of 0.06 μg/mL. The AF, ESRD, DM, and sepsis patient samples had a mean endogenous GAG concentration of 0.55, 0.72, 0.92, and 0.94 μg/mL, respectively. The levels of endogenous GAGs were highest in cancer, LD, and PE patient plasma samples with a mean concentration of 1.95, 2.78, and 2.83 μg/mL, respectively. Heparinase I degradation resulted in a decline in GAG levels in plasma samples. These results clearly show that detectable Heparin Red sensitive endogenous GAGs are present in circulating plasma at varying levels in various patient groups. Additional studies are necessary to understand this complex pathophysiology.
Introduction
Vascular endothelium has a major role in circulatory homeostasis. It is comprised of a single layer of squamous cells lining the entire circulatory system. As it forms a physical barrier for blood cells, they regulate the vascular tone and control intravascular coagulation. The vascular endothelium, also regulates the leukocyte and platelet interactions, smooth muscle cell proliferation, and modulates the vascular inflammation by releasing hormones and other soluble mediators such as cytokines. 1 Endothelial glycocalyx which is expressed on the endothelial cells, is a mixture of glycosaminoglycans (GAGs) attached to proteins. 2 Studies have shown that endothelial damage can cause glycocalyx dysfunction causing enzymatic degradation (shedding) of its components, which further leads to their detection in the circulating bloodstream. 3
GAGs are a family of structurally complex heteropolysaccharides comprised of alternating hexosamines with uronic acid or glucuronic acid residues that include heparin and heparan sulfate (HS), chondroitin sulfate (CS), dermatan sulfate (DS), keratan sulfate (KS), and hyaluronic acid (HA). 4 Biological functions of glycans can be divided into 3 broad categories: (1) structural contributions (eg, cell wall and extracellular matrix synthesis, protein folding, and function), (2) energy metabolism (eg, carbon resources for energy and storage), and (3) information carriers (eg, signal transduction, enzyme inhibition, growth factor activation). 5 The surface of all types of cells is covered with a dense layer of GAGs, in nature. 6 They can be found intracellularly, usually within secretory granules. They are the major component of the extracellular matrix. However, the density, chemical structure or composition of polysaccharide chains, and protein type or content differentiate significantly in different cell types and various species.
The presence of circulating GAGs in human plasma has been demonstrated in previous studies by several investigators.7–9 In these studies, while CS is shown as the main circulating GAGs in normal human plasma (NHP), HS, DS, and HA have shown in very low levels.10–12 Alterations in the levels of endogenous GAGs have been attributed to several disease processes ranging from mucopolysaccharidosis to complex diseases such as collagen vascular disease, sepsis, and cancer.13–15 For this reason, the detection of endogenous GAGs in biological samples may evolve to be a group of promising biomarkers for the early detection of different disease states. Endogenous GAGs are characterized utilizing advanced methods in bleeding patients by Linhardt et al.16,17 Additionally, heparin-like substances have been extracted from human hemangioma and reported in an earlier publication. However, GAG composition and length may vary in different tissues due to different enzymatic processes, different pathophysiological insults, or even different mechanisms of glycocalyx degradation which eventually may affect characteristic GAG patterns. 18
While the endogenous GAGs in circulating blood may originate from the glycocalyx matrices at the endothelial surfaces, organ-specific GAGs originated from liver, kidney, heart, lungs, and brain may also be present in different pathologic states.19,20 These GAGs are of heterogeneous origin and have not been fully characterized. A diagrammatic illustration of the release of endogenous GAGs is depicted in Figure 1. Matrix-degrading enzymes and damage to endothelium may result in the release of GAGs in the blood. Various pathologic conditions including deep vein thrombosis, PE, vasculitis, and ruptured atherosclerotic plaque may lead to the release of GAGs. 20 The role of GAGs in infectious disease and related disorders has been reported. These findings may be relevant to the role of these agents in different diseases. 21 On the other hand, tissue factor and thrombin-dependent formation of clots and related products may facilitate additional augmentation of the release of these GAGs. Tissue damage and necrosis also contribute to the activation of platelets. These processes may result in the release of platelet factor 4 and other polyanions which can be complex with endogenous GAGs. Anti-heparin platelet factor 4 antibodies have been reported in patient groups who are heparin naive, and these may be the result of the complexation of endogenous GAGs. Endogenous GAGs have not been extensively studied in biologic fluids such as plasma. While the presence of endogenous GAGs has been reported there are no definitive reports on the quantification of these agents in plasma or other biological fluids. 22 With the availability of newer methods, it may be possible to quantify these GAGs as absolute levels and further characterization of molecular structural features.

Thrombo-inflammatory response in the mediation of the generation of the circulating glycosaminoglycans (GAGs) and anti-GAG PF4 antibodies. Endogenous damage of the endothelial lining amplifies the generation of GAGs which may be responsible for measurable levels of heparin-related GAGs. These GAGs may be complex with PF4 and generate antibodies.
Several qualitative and quantitative methods such as dye-spectrometry, liquid chromatography, polyacrylamide gel electrophoresis, enzyme-linked immunosorbent-assay, mass spectrometry, and nuclear magnetic resonance have been developed for the measurement of GAGs in biological samples and evaluation of its role in the biological process.13,23 However, very few readily available techniques exist to detect and measure GAGs, which are difficult to standardize and expensive to run. Heparin Red probe is a novel fluorescence assay that quantifies heparins (including unfractionated heparin, low molecular weight heparin, and non-anticoagulant heparins) and other polyanionic sulfated GAGs with a sensitivity range of 0 to 6 µg/mL. 24 Heparin Red is a polyamine derivative of a red-emissive perylene diimide fluorophore. It forms a supramolecular complex with the target, with aggregation of the probe molecules at the heparin template and contact quenching of fluorescence. The strong binding of the polycationic probe to polyanionic GAG appears to be controlled by electrostatic and noncovalent bonds between aromatic rings. The probe enables direct detection of heparins and other sulfated GAGs by a simple mix-and-read microplate assay which is independent of any anticoagulant activity. However, it is prone to interference with other endogenous polyanions (eg, HS, hyper-sulfated CS, nucleic acids, polyphosphates, lipid-A) which usually are not present in blood plasma at significant concentrations.25–30
Heparin and HS can be digested by heparinases, a group of bacterial lyases that are widely used as tools for processing and analyzing these polysaccharides. 31 Heparinases degrade heparin and HS GAGs through an eliminative mechanism. Heparinase-I from Flavobacterium heparinum is highly specific on heparin and has no activity against CS and KS. It cleaves a hexosamine-iduronic linkage with an O-sulfated-L-iduronic acid residue. Heparinase-II is active against heparin and HS-like regions of GAGs and cleaves hexosamine-iduronic/glucuronic acid linkages. Heparinase-III presents a substrate specificity for hexosamine–glucuronic acid disaccharide sequences. 32 Thus, we tried to rule out the effects of possible therapeutic heparin contamination in plasma samples by using Heparinase-I.
In this study, we sought to measure the levels of endogenous GAGs before and after heparinase-I degradation in various plasma samples in comparison to healthy controls, in order to determine the influence of glycocalyx degradation in the complex pathophysiology of various disease conditions. A highly sensitive fluorescence quenching method capable of detecting submicrogram levels of GAGs was utilized.
Materials and Methods
Blood Samples
De-identified whole blood samples were obtained from various patient groups and retrieved from clinical laboratories of Loyola University Hospital (Maywood, Illinois) under an Institutional Review Board-approved protocol. Samples were collected in 3.8% (0.109 mol/L) sodium citrate tubes and processed for platelet-poor plasma, and stored at − 80 °C prior to analysis. NHP samples from healthy, nonsmoking, adults, aged 19 to 53, were purchased from a commercially available source (George King Biomedical, Overland Park, Kansas).
Measurement of Endogenous GAGs in Different Populations
The Heparin Red method based on the fluorescence quenching method was used. The reagents were obtained from Red Probes (Munster, Germany). For the determination of endogenous GAG concentrations in each plasma sample, the protocol of the provider for a 96-well microplate assay was followed. A mixture of 10 000 μL Heparin Red solution and 90 μL Enhancer solution was freshly prepared. A total of 20 μL of the patient or healthy control sample were pipetted into a microplate well, followed by 80 μL of the Heparin Red–Enhancer mixture. Immediately after mixing, the microplate was introduced in the fluorescence reader (Biotek Cytation-5 microplate reader) and fluorescence was recorded within 1 min.
The absolute levels of endogenous GAGs were quantified in reference to a calibration curve prepared by using porcine mucosal heparin (Medefil, Inc.). Medefil heparin was obtained from Medefil Inc. (Glendale Heights, Illinois) and is a standard unfractionated heparin. The calibration curves were prepared by manufacturers’ guideline.24,25
Measurement of Endogenous GAGs After Heparinase I Degradation
In order to measure the endogenous GAG content in plasma samples after heparinase-I degradation Heparinase-I from Flavobacterium heparinum (Sigma-Aldrich) was used. For this purpose, an aliquot of 70 μL of each sample was added to an Eppendorf tube along with 10 μL of CaCl2, and 10 μL (10 U) of heparinase-I. An additional 10 μL of saline was added for a total volume of 100 μL. The solutions were mixed, incubated for 30 min at 37 °C, and then placed in a 100 °C hot water bath for 3 min to terminate the reaction. The final heparinase-I concentration was 1 U/mL. After incubation, endogenous GAG content of each plasma sample was measured by Heparin Red assay.
Statistical Analysis
Circulating levels of endogenous GAGs in patient plasma were compared to NHP. Descriptive statistics were calculated utilizing GraphPad Prism software. Statistical differences between PE groups and normal controls were evaluated utilizing nonparametric Mann-Whitney U, Student t-tests, and Kruskal-Wallis ANOVA. A p-value of <.05 was considered statistically significant.
Results
As shown in Table 1, NHP was found to have relatively low levels of endogenous GAGs with a mean concentration of 0.06 μg/mL. Atrial fibrillation (AF) patient samples had a mean endogenous GAG concentration of 0.55 μg/mL. The end-stage-renal-disease (ESRD) patients had a mean endogenous GAG concentration of 0.72 μg/mL. The diabetes mellitus (DM) and sepsis patients had similar levels of endogenous GAGs with a mean concentration of 0.92 μg/mL and 0.94 μg/mL, respectively. The cancer patients had a mean endogenous GAG concentration of 1.95 μg/mL. The levels of endogenous GAGs were highest in liver disease (LD) and pulmonary embolism (PE) patient plasma samples with a mean concentration of 2.78 μg/mL and 2.83 μg/mL, respectively. All the patient samples were significantly higher in comparison to the NHP samples (Figure 2).

The levels of endogenous glycosaminoglycans in different plasma samples. All the patient samples were higher in comparison to the normal human plasma (NHP) samples.
Concentrations of Endogenous Glycosaminoglycans in Different Plasma Samples.

After incubation with heparinase-I, NHP samples have shown a mean endogenous GAG concentration of 0.05 μg/mL (16.67%-change). The mean endogenous GAG concentration of AF patient samples was 0.42 μg/mL (23.64%-change), ESRD patient samples was 0.36 μg/mL (50.00%-change), DM patient samples was 0.73 μg/mL (20.65%-change), Sepsis patient samples was 0.46 μg/mL (51.06%-change), cancer patient samples was 1.62 μg/mL (16.92%-change), LD patient samples was 2.62 μg/mL (5.76%-change), and PE patient samples was 1.46 μg/mL (51.59%-change). The results of endogenous GAG concentrations after heparinase-I degradation are represented in Table 2. The patient samples were still significantly higher in comparison to the NHP samples after Heparinase-I degradation (Figure 3).
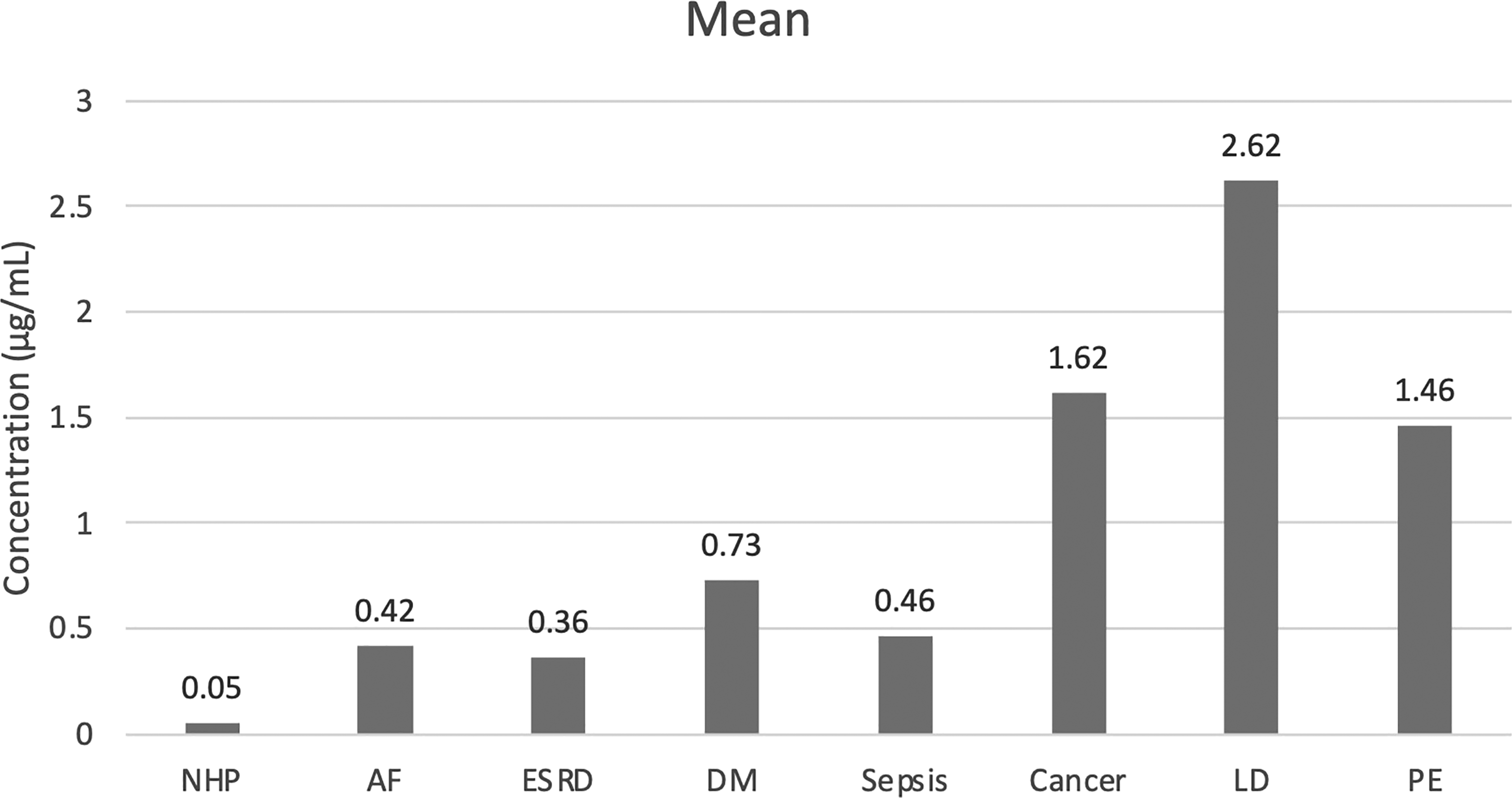
The levels of endogenous glycosaminoglycans after heparinase-I degradation. The patient samples were still significantly higher in comparison to the normal human plasma (NHP) samples after heparinase-I degradation.
Concentrations of Endogenous Glycosaminoglycans After Heparinase-I Degradation.

Discussion
Posttranslational modification of proteins enhances their functional heterogeneity and is important for many biological processes. In eukaryotic cells, proteins are co-translationally translocated into the endoplasmic reticulum (ER) and Golgi apparatus during their synthesis. 33 In ER/Golgi complex, these proteins go through various posttranslational modifications in order to become functional proteins. These modifications are mostly glycosylation processes and comprised of N-glycosylation, O-glycosylation, and other processes such as the production of glycosylphosphatidylinositol (GPI) anchor proteins, various glycoproteins, and glycolipids. 34 N-glycosylation refers to the addition of saccharide chains to the nitrogen of asparagine residues (N-linked) and begins in the ER. O-glycosylation refers to the addition of saccharide chains to the hydroxyl group of either serine or threonine residues of the protein, and it occurs exclusively in the Golgi apparatus. The enzymes involved in each of these modification pathways are distinct and include different glycosyltransferase, glycosidase, and glycan-processing enzymes. 35 Following these glycosylation processes the synthesized proteins are transferred to their functional locations such as the cell surface for membrane proteins, intracellular vesicles for secreted proteins, or lysosomes for lysosomal enzyme proteins. For this reason, endogenous GAGs can be located in the intracellular cytoplasms, the cell membrane, or in the extracellular matrix (ECM). The endothelial glycocalyx consists of a two-layered structure: an inner dense matrix layer associated with membrane-attached glycoproteins that forms a primary selective barrier to plasma macromolecules, and an outer less dense layer composed mainly of GAGs and plasma proteins that extends one or more micrometers into the vessel's lumen and supports continuous blood cell movement while restricting inflammatory cell access to the endothelial surface.
Numerous studies have shown that AF is associated with endothelial dysfunction through multiple mechanisms. These mechanisms can be listed as; altered hemodynamics, increased shear stress on endothelial cells, reduced nitric oxide (NO) bioavailability, increased oxidative stress and inflammation, renin-angiotensin dysregulation, and intracellular Ca+ 2 overload. 36 Oxidative stress-related pathophysiological changes (such as increased NADPH and xanthine oxidase activity, deletion of mitochondrial deoxyribonucleic acid) have been consistently demonstrated in AF patients. 37 Several studies utilized flow-mediated-dilation (FMD) which is a measure of peripheral artery endothelial function to demonstrate the endothelial dysfunction in AF. In these studies, FMD was shown to be impaired in AF patients in comparison to healthy controls and was also related to the severity of AF. 38 Interestingly, the impaired FMD in AF patients was able to be restored following cardioversion and catheter ablation, suggesting that the impaired endothelial dysfunction is linked to AF itself and is not simply the consequence of other cardiovascular risk factors. 39 Furthermore, worse peripheral microvascular endothelial dysfunction was associated with a higher recurrence of AF following catheter ablation. 40 The other measures of peripheral endothelial dysfunction (such as the peripheral arterial tonometry, and brachial artery diameter studies) were also supportive of these findings.36–38 Additionally, while the levels of NO were significantly reduced in AF patients, the levels of asymmetric dimethylarginine (ADMA), Von Willebrand factor (vWF), circulating microparticles, C-reactive protein (CRP), interleukin-6 (IL-6), adhesion molecules (such as E-selectin and VCAM-1), and circulating extracellular vesicles have shown to be increased in patients with AF, suggesting the possible contribution of oxidative stress and inflammation to endothelial dysfunction in development and progression of AF. 41 Additionally, increased levels of clotting factors (tissue factor, factor VIII, fibrinogen), platelet activation markers (platelet factor 4, β-thrombomodulin, P-selectin), and fibrinolysis markers (D-dimer, tissue plasminogen activator, plasminogen activator inhibitor-1) are observed in AF which is associated with increased frequency of thromboembolic events in AF patients. 42 In our current study, the levels of endogenous GAGs were elevated in AF patient plasma, compared to NHP, which is possibly a reflection of endothelial damage and glycocalyx degradation leading to the development of AF in these patient groups.
Similar to AF, endothelial dysfunction has also been reported in ESRD patients. Atherosclerotic cardiovascular complications (such as myocardial infarction, stroke, and peripheral arterial disease) are the leading causes of mortality and morbidity in ESRD, and endothelial dysfunction seems to be the starting point of these adverse outcomes. 43 Regarding this, decreased levels of NO and increased levels of ADMA concentrations have been observed in uremic ESRD patients suggesting a high level of oxidative stress in these patients.44,45 Additionally, biomarkers of endothelium and inflammation such as TNFα, IL-1, IL-6, monocyte-chemoattractant-protein-1 (MCP-1) CRP, vWF, selectins, VCAM-1, and ICAM-1 are also reported in increased levels in ESRD patients. 46 These findings were partly explained by induction of NF-kB pathway which triggers the synthesis of adhesion molecules, cytokines and chemokines, coagulation factors, and vasoactive mediators. Furthermore, the plasma indoxyl sulfate is a uremic toxin that accumulates in ESRD patients, causing endothelial damage. 47 The erythropoietin (EPO) which has vasculo-protective properties is reduced in ESRD patients. 48 It's been shown that the levels of advanced glycosylated end products (AGE) are also increased in ESRD patients. These compounds are known to promote inflammation, oxidative stress, and endothelial dysfunction, leading to adverse outcomes that are observed in ESRD. 49 In our current study, although we observed a 50% reduction after heparinase-I degradation, the levels of endogenous GAGs were much higher than the NHP which can be explained as a possible sign of endothelial dysfunction and glycocalyx shedding that is observed in these patients. It is proposed that the high percentage of decrease in endogenous GAG levels after heparinase-I administration is mostly related to heparin contamination due to the high frequency of therapeutic heparin treatment that is used in these patients.
A large body of evidence links endothelial dysfunction to DM. Several observational studies have shown reduced endothelium-dependent vasodilation in diabetic patients. 50 Moreover, circulating levels of endothelial biomarkers (eg, TF, vWF, thrombomodulin, selectins, type IV collagen, VCAM-1, ICAM-1, Endothelin-1) were found to be elevated DM. 51 Basically, the mechanism of endothelial damage was linked to blood glucose fluctuations, chronic hyperglycemia and selective insulin resistance with hyperinsulinemia in diabetic patients. 52 Regarding this, chronic hyperglycemia and blood glucose fluctuations result in increasing the entry of glucose into different metabolic pathways (eg, polyol, hexosamine, protein-kinase-C, and AGE pathways), insulin resistance and hyperinsulinemia activates PI3-kinase/Akt and MAPK/ERK pathways leading to generation of reactive oxygen species (ROS) and decreased NO synthesis in diabetic patients. Furthermore, increased levels of inflammatory biomarkers (eg, CRP, TNFα, IL-1, IL-6, and MCP-1) and activation of coagulation factors (eg, fibrinogen, PAI-1, t-PA, FVII, FVIII, FXI, and FXII) suggest a proinflammatory and prothrombotic state in DM. 53 In our current study, the levels of endogenous GAGs were constantly elevated in DM patient plasma compared to NHP, showing a possible link to ongoing endothelial damage and glycocalyx degradation in these patients.
Sepsis induces rapid and profound changes in coagulation system and endothelial function. Endothelial activation and dysfunction are critical determinants of the host response and is the main component of the complex pathophysiology of this syndrome. 54 Sepsis is characterized as a catabolic state of degradation of proteins, lipids, and carbohydrates. It has been shown that the degradation of the endothelial glycocalyx is carried out by sheddases, disintegrin and metalloproteinases (ADAMs), matrix metalloproteinases (MMPs), heparanase-I, and hyaluronidases in sepsis. 55 Moreover, the analysis of LPS-induced endothelial dysfunction has shown increased glycan production (together with fatty acid metabolism) to counterbalance glycocalyx loss in endothelial cells in septic patients. 56 As expected, the levels of CRP, TNFα, IL-1, and IL-6 are increased in septic patients. 57 VWF, Angiopoietin-2 (Ang-2), endocan, ET-1, selectins, and other adhesion molecules, VEGF and its soluble VEGF-receptor-1 (Flt-1), and PDGF have shown in elevated levels and were correlated with organ dysfunction, severity, and mortality of sepsis.58,59 Furthermore, the levels of uPA, tPA, and PAI-1 which are elevated in sepsis, also provide information about endothelial cells, because, upon stimulation of endothelial cells, levels of these fibrinolytic proteins increase in the circulation. 59 In our current study, our results have shown elevated levels of endogenous GAGs in sepsis patients compared to NHP supporting previous reports of endothelial damage and increased glycocalyx turnover in sepsis patients. Here again, the high percentage of the decline of endogenous GAG levels after heparinase-I administration might either be related to contamination of therapeutic heparin treatment that is used in these patients or may be the result of endogenous heparin release from platelets and microparticles.
Cancer cells are also covered with a glycocalyx layer which is located between the extracellular cell matrix and cell membrane of the cancer cells depending on the type of the tissue and cell and their location in organ systems. 60 The interactions of glycocalyx with surrounding tissue may affect tumor growth, cancer cell survival, cancer cell adhesion and migration, angiogenesis, invasion, and metastasis of cancer cells. 61 Tumor cells exhibit bulky amounts of the glycocalyx. 62 The bulky glycocalyx causes an increase in the number and activity of membrane-bound and ECM-bound signaling molecules which promote carcinogenesis. 63 Furthermore, abundant glycosylation and differential expression of structural GAGs such as syndecans, HA, and HS in glycocalyx together with increased MMP activity have been shown in several cancer studies supporting their role in the complex pathophysiology of carcinogenesis. 64 Interestingly, reduced vascular endothelial glycocalyx thickness has been shown in cancer patients in comparison to healthy controls which was independent of traditional cardiovascular risk factors and anticancer therapy in one study. 65 In our current study, our results have shown a constant elevation in the levels of endogenous GAGs in comparison to healthy controls. Considering the fact that the studies regarding glycocalyx and circulating GAGs in cancer patients are scarce, these results require further investigation.
The liver receives about 25% of the cardiac output from the portal vein and the hepatic artery. The liver microcirculation is comprised of hepatic sinusoids, which are lined by a thin fenestrated endothelium without a basement membrane. 66 Liver sinusoidal endothelial cells (LSECs) and Kupffer cells (KCs) represent the predominant nonparenchymal cell types of the hepatic sinusoid, in addition to inflammatory cells such as dendritic cells (DCs). The LSECs, DCs, hepatic stellate cells (HSCs), and KCs have been shown to recognize antigens and play a key role in the liver immune response. Together with KCs the LSECs play a key role in host defense mechanism and blood flow regulation and represent a major target for injury during LD.67,68 During acute and chronic liver injury, LSECs become “capillarized,” a term associated with the loss of fenestrae and development of a basement membrane, similar to common capillary endothelium. 69 Furthermore, they lose their protective properties, acquiring vasoconstrictor, pro-inflammatory and prothrombotic functions. Additionally, loss of fenestrae and basement membrane deposition impedes the appropriate oxygenation of hepatocytes, resulting in apoptosis and necrosis and the secretion of damage-associated-molecular-patterns (DAMPs). 70 Following this, LSEC-derived factors, produce an excess of extracellular matrix and promote fibrosis development. It has been shown that similar to vascular endothelium, hepatic sinusoids also have a glycocalyx layer which is comprised of GAGs. 71 This layer forms large and irregularly anastomosing structures and does not occlude the open fenestrations of the sinusoid. Furthermore, the glycocalyx around the endothelial cells, is not only on the luminal side but also on the side facing the space of Disse. Alteration of glycosylation properties of liver GAGs has been shown in various LDs including cirrhosis, fatty LD, viral hepatitis, and hepatocellular carcinoma. 72 The current study is consistent with previous reports suggesting an elevation in the levels of endogenous GAGs in the circulation of LD patients.
The current knowledge on the contribution of vascular endothelium and glycocalyx degradation on the pathophysiologic mechanisms of PE development is limited. An impaired flow-mediated dilation has been shown in 2 recent studies in PE patients which suggests the possible involvement of endothelial dysfunction in PE development.73,74 Syndecan-1 which is a glycocalyx degradation biomarker, was elevated in PE patients in another study. 75 In a recent analysis performed by our group, has shown that the levels of endogenous GAGs were elevated in PE patients, and this was also associated with PE severity and mortality.76,77 In the current study, our findings are similar to a previous report showing elevated levels of endogenous GAGs in PE patient plasma compared to NHP, both before and after heparinase-I degradation which is likely the result of endothelial damage and glycocalyx degradation leading to PE development in these patients. The high percentage of decrease in endogenous GAG levels after heparinase-I administration might be the result of heparin contamination of plasma samples in which therapeutic heparin is the main treatment option in this patient group. The observed higher levels of endogenous GAGs detected in this cohort may likely be due to the presence of therapeutic heparin which is routinely used in the management of PE. In our studies, we demonstrated a significant reduction of Heparin Red sensitive GAGs after heparin digestion suggesting the presence of heparin. To further characterize the presence of other GAGs the removal of heparin from these samples by chemical or enzymatic methods and subsequent structural characterization of the residual GAGs will provide detailed information on the composition of these polymers in PE.
Because of the methodological issues, only limited data is available on the quantitation and structural properties of endogenous GAGs. The current studies have underscored that measurable amounts of GAGs are detectable in plasma obtained from different patient groups. The heparin red detectable GAGs may have structural diversity due to their origin.78,79 The heparin red method provides an absolute method for the quantitation of GAGs and further studies on structural characterization are needed. The Heparin Red method may also be sensitive to synthetic sulfated polymers such as dextran sulfate, synthetic heparins, and heparinoids, and over-sulfated CS because of the polyanionic nature of these agents. Moreover, the isolation and characterization of plasmatic GAGs require step-wise approaches including purification and molecular characterization using chromatographic and mass spectrometric methods. Because of the low levels of circulating GAGs, sensitive methods are needed to profile these important polymers which may have different profiles in specific pathologic states.
Study Limitations
In this study, the patient demographics and baseline clinical characteristics were not included because of the de-identified nature of the blood samples. This study is based on single sample analysis and due to logistic reasons, follow-up samples were not collected and analyzed. Future studies should consider collecting sequential samples in the follow-up analysis. Additionally, we were not able to consider the previous anticoagulant history and the type of anticoagulant used during sample collection. For this study, it is important to know the history of heparinization in terms of the type of heparin and mode of administration. We used heparinase-I to rule out possible heparin contamination in plasma samples. The Heparin Red method has limited sensitivity and is capable of detecting submicrogram levels of endogenous GAGs. It has limited sensitivity to polyanions with decreased charge density, lower molecular weights, and circulating levels below 500 ng/mL. In the current study, we did not compare the Heparin Red method with other methods which are currently used for the quantification and structural characterization of GAGs such as dye-binding methods, mass spectrometry, and chromatographic methods which would have provided supportive data and the relevance of Heparin Red quantifiable GAGs to these methods. Despite these limitations, this method is capable of detecting measurable levels of global GAGs in the plasma samples obtained from different pathologic states.
Conclusion
This study has been reported on the application of a method based on the utilization of a fluorescence probe for the quantification of heparin-related GAGs in plasma samples obtained from various pathological states. This method offered a simple and rapid assay for measuring global GAG levels in plasma samples. Our results have shown that the levels of endogenous GAGs are elevated in all the tested plasma samples in comparison to healthy controls, which proves a clear association between endothelial damage and glycocalyx degradation in the complex pathophysiology of these different disease states. The vascular surface composition includes proteoglycans regardless of the circulatory bed and any damage to the surface results in the release of GAGs which can be detected by using the Heparin Red method. In addition to this, the results were consistent both before and after heparinase-I degradation, which rules out the possible heparin interactions in these plasma samples. In routine clinical practice, different biomarkers are commonly used to detect the course, prognosis, and adverse outcomes of these different disease states, but direct markers reflecting endothelial damage are not commonly available. In this study, we used a novel fluorescence assay which is a simple easy-to-use method to detect endogenous GAGs in circulation. To further validate the utility of this method for the rapid detection of GAG levels, additional well-designed studies in clinically proven pathological states are needed. Additionally, the relevance of structural features of GAGs and variations in their circulating level with reference to endothelial damage and matrix remodeling may provide additional insight into the pathophysiologic role of GAGs in health and disease. The simplicity, rapid turn-around time, and cost to perform quantification of GAGs by the Heparin Red method may offer an advantage for the mass screening of patient cohorts to quantify these agents and provide a platform to characterize their structure.
Footnotes
Acknowledgments
The authors are thankful to the staff of the clinical laboratories for the facilitation of the collection of blood samples. A special thanks to Professor Kraemer of the University of Heidelberg for providing the generous gift of Heparin Red agents used in this study. The authors also acknowledge the helpful advice and guidance from Professor Robert Linhardt Rensselaer Polytechnic Institute (Troy, NY) for the study. We are thankful to Dr Seth Robia and Dr Alain Heroux for their support. We are also grateful to Dr Lowell Steen for providing helpful advice and encouragement to carry out this research program. The skillful assistance of Ms. Erin Healy-Erickson in the preparation of this manuscript is gratefully acknowledged.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.