
Abstract
Introduction
Thalassemia is a kind of hereditary hemolytic blood disease, which is mainly prevalent in tropical and subtropical regions of the world.1,2 Thalassemia is the most common autosomal recessive genetic disease, and its pathogenic mechanism is the imbalance of human globin expression, so it is also known as globin synthesis disorder. There are 350 million thalassemia carriers in the world, and more than 300 000 newborns are born with sickle cell anemia or thalassemia each year.3,4 Southern China has a high incidence of thalassemia, and the population carrier rate is 3-24%. This disease has different carrier rates in different regions of China with strong genetic heterogeneity and ethnic and regional differences. Previous study showed that thalassemia occurred mostly in Guangxi, Yunnan, Hainan and Guangdong. 5 –9 According to the epidemiological research data reported by Xu in 2004, 5 the total carrier rate of thalassemia in Guangdong was as high as 11.07%, and the rate of α-thalassemia, β-thalassemia, α compound β thalassemia was 8.53%, 2.54% and 0.26%, respectively. In 2014, Yin figured out that the carrying rate of thalassemia was highest in western and mountainous areas of Guangdong, and lowest in Chaoshan area. 10 With the development of national economy, previous data on thalassemia needs to be updated. As an autosomal recessive genetic disease, the majority of thalassemia carriers have no clinical symptoms. The hematological data of α-thalassemia silent (-α3.7/αα, -α4.2/αα and αWSα/αα) are usually in the normal range, but sometimes microcyte and/or hypochromia may appear. The life span, growth and development of thalassemia carriers are not significantly different from those of normal people, but the children of carrier couples have a one-quarter chance of suffering from severe hemolytic anemia symptoms. At present, the only way to cure thalassemia is bone marrow stem cell transplantation. Prenatal screening and diagnosis can be used to identify the thalassemia carriers, so that the pregnancy can be terminated in time to prevent the child’s birth.
There are more than 130 types of mutations in α-thalassemia (more than 40 deletion and more than 90 non-deletion types) 11 and more than 300 types of mutations in β-thalassemia. 12 ,13 Nowadays, the conventional molecular diagnostic techniques for thalassemia are Gap-PCR and RDB-PCR; rare type Gap-PCR, Sanger sequencing and multiplex ligation-dependent probe amplification (MLPA) are used for detection when special mutation types are encountered. Studying the latest gene mutation spectrum and prevalence of thalassemia, exploring the distribution and variation types of α-thalassemia and β-thalassemia, can provide a scientific theoretical basis for the diagnosis, treatment and prevention of thalassemia. It also provides accurate and timely help during prenatal diagnosis or reproductive genetic counseling.
Materials and Methods
Patients and Samples
From January 1, 2015 to December 31, 2020, a total of 31 512 peripheral blood and 3828 fetal samples were collected from the Reproductive Medicine Center and Prenatal Diagnosis Department in The Third Affiliated Hospital of Guangzhou Medical University. Our hospital is also a superior referral hospital, so all the peripheral blood samples (couples and probands) of Reproductive Medicine Center and Prenatal Diagnosis Department come from different cities in Guangdong province such as Guangzhou, Foshan, Qingyuan, Yunfu, Shaoguan, Shanwei, etc The proportion of each region is 5.40% in eastern GuangDong, 9.09% in western GuangDong, 11.79% in northern GuangDong, and 73.72% in central GuangDong (Pearl River Delta). The reproductive center conducted a general survey of thalassemia genes in all routine patients (including couples and proband), and the prenatal diagnosis department did prenatal diagnoses on the 3828 fetuses if their parents carried the same type of thalassemia and the fetus has a 1/4 chance of having moderate or major thalassemia. All the patients signed the informed consent forms.
Hematological Studies
Hematological results of EDTA-anticoagulant-treated peripheral blood were analyzed by automated hematology analyzer (Sysmex, Japan), and hemoglobin components and levels were performed by using capillary electrophoresis (CE; Serbia, Paris, France).
Clinical Genotype Phenotype Definition
α-thalassemia silent (TS) are defined as follows: -α3.7/αα, -α4.2/αα and ααWS/αα; Thalassemia trait (TT): ααCS/αα, ααQS/αα, --SEA /αα, -α4.2/-α4.2, -α4.2/-α3.7, -α3.7/-α3.7,-α4.2/ααWS, -α3.7/ααWS, ααWS/ααWS, ααCS/ααWS, ααQS/ααWS, --SEA /ααWS; Thalassemia
Intermedia (TI): -α3.7/ααQS, -α3.7/ααCS, -α4.2/ααQS, -α4.2/ααCS, ααCS/ααQS, ααCS/ααCS, ααQS /ααQS, --SEA /-α3.7, --SEA/-α4.2, --SEA/ααQS, --SEA/ααCS; Thalassemia Major (TM): --SEA /--SEA. β-thalassemia genotype are defined as follows: β0 (absence of HBB gene expression): HBB:c.-78A>G, HBB:c.2T>G, HBB:c.45_46insG, HBB:c.52A>T, HBB:c.84_85insC, HBB:c.92+1G>T, HBB:c.113G>A, c.124_127delTTCT, HBB:c.130G>T, HBB:c.216_217insA, Chinese del (NC_000011.9:g.5191148_5270051del); β+ (reduce of HBB gene expression): HBB:c.-140C>T, HBB:c.-79A>G, HBB:c.-50A>C, HBB:c.79G>A, HBB:c.316-197C>T, SEA-HPFH (NC_000011.9:g.5222878_5250288del). Heterozygotes for β-thalassemia (β0/βN, β+/βN) usually manifest as thalassemia trait (TT), homozygotes or double heterozygotes for β-thalassemia (β0/β+, β0/β0) usually manifest as thalassemia major (TM), while thalassemia intermedia (TI) are more complex which are often affected by factors such as α-thalassemia genotype and modifier genes.
Molecular Analysis
DNA was extracted from peripheral blood, chorionic villi, amniotic fluid or cord blood by using Qiagen DNA kit (Qiagen; Hilden, Germany). For common genotyping, Gap-PCR was used for 3 α-globin gene deletions, and reverse dot blot (RDB) was used for 3 non-deletion α-thalassemia and 17 non-deletion β-thalassemia. Specific Gap-PCR, Gel electrophoresis and Sanger sequencing were performed for the patients who were highly suspected of thalassemia.
Statistical Analysis
Hematological parameters among different genotype groups were compared by Student’s t-test. A P-value less than .05 was considered a significant difference.
Results
19.48% (6137/31 512) cases were diagnosed as thalassemia, of which 4749 (15.07%) cases were α-thalassemia, 1196 (3.80%) cases β-thalassemia and 192 (0.61%) cases co-inheritance of α- and β-thalassemia. For prenatal samples, 82.55% (3160/3828) cases were diagnosed as thalassemia, in which 2021 (52.80%) were α-thalassemia, 997 (26.05%) were β-thalassemia and 142 (3.71%) were co-inheritance of α- and β-thalassemia. The prevalence of peripheral blood and prenatal samples and the composition ratio of different clinical phenotypes were shown in Figure 1.

The prevalence of peripheral blood and prenatal samples and the composition ratio of different thalassemia. A and E represent the prevalence of thalassemia in the peripheral blood samples and prenatal samples, respectively; B, C, D respectively represent the proportion of different clinical phenotypes in each type of thalassemia in postnatal samples; F, G, H respectively represent the proportion of different clinical phenotypes in each type of thalassemia in prenatal samples. #TS: Thalassemia silent; TT: Thalassemia trait; TI: Thalassemia Intermedia; TM: Thalassemia Major.
Hematological Results
1000 females and 1000 males, who were of childbearing age between 18 and 45, routine population in reproductive centers with normal blood routine and hemoglobin electrophoresis were involved in the normal group, and the common α-thalassemia hematological data were shown in Table 1. Compared to the normal group, only --SEA/αα had a significant difference. -α3.7/αα decreased in MCV, MCH and HbA2 level; -α4.2/αα reduced in MCV and MCH level; ααCS/αα was lower in Hb A2 level; ααQS/αα decreased in HGB and MCV level. The results would also be affected by gender. All the data of β-thalassemia patients had a remarkable different compared to the normal group. For co-inheritance of common α- and β-thalassemia, the Hb A2 level of all thalassemia groups increased significantly, and only thalassemia trait combined β0 patients decreased dramatically in MCV and MCH level. The hematological data of different Hb H diseases was shown in Table 2, and the hematological and genotype data of β-thalassemia intermedia and major patients was listed in Table 3.
Hematological Data of Common α-Thalassemia, β-Thalassemia and α- + β-Thalassemia.

#People of childbearing age: 18-45 years old, compared with the normal group, * means P < .05. TS: Thalassemia Silent; TT: Thalassemia Trait.
Hematological Data of Hb H Disease
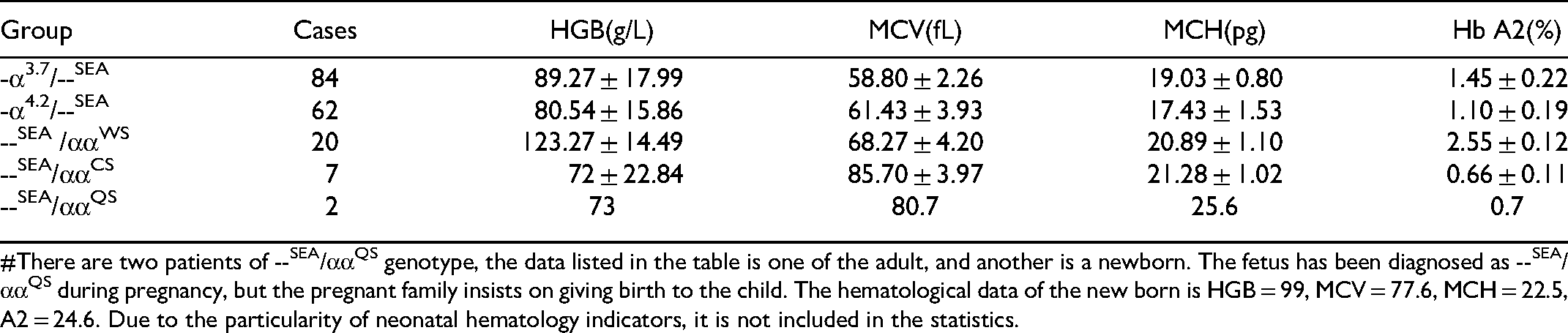
#There are two patients of --SEA/ααQS genotype, the data listed in the table is one of the adult, and another is a newborn. The fetus has been diagnosed as --SEA/ααQS during pregnancy, but the pregnant family insists on giving birth to the child. The hematological data of the new born is HGB = 99, MCV = 77.6, MCH = 22.5, A2 = 24.6. Due to the particularity of neonatal hematology indicators, it is not included in the statistics.
Hematological and Genotype Data of β-Thalassemia Tntermedia and Major Patients

#Thalassemia Trait:TT; TI: Thalassemia Intermedia; TM: Thalassemia Major. βCD27−28/β−28 and βCD71−72/βE are pregnant women in high-risk obstetrics, other 4 cases are postpartum children/probands. Hematological parameters are not available for intermedia and major antenatal samples due to labor induction.
Clinical Phenotyping
In 4749 peripheral blood α-thalassemia cases, there were 1722 (36.26%) Thalassemia Silent (TS); 2851 (60.03%) Thalassemia Trait (TT); 175 (3.69%) Thalassemia Intermedia (TI) and 1 (0.02%) Thalassemia Major (TM). In 2021 prenatal α-thalassemia cases, there were 458 (22.66%) TS; 1053 (52.10%) TT; 144 (7.13%) TI and 366 (18.11%) TM.
In 1196 peripheral blood β-thalassemia cases, there were 1159 (96.91%) TT; 22 (1.84%) TI and 15(1.25%) TM. In 997 prenatal β-thalassemia cases, there were 721 (72.32%) TT; 27 (2.73%) TI, 249 (24.97%) TM.
For co-inheritance of α- and β-thalassemia, 192 blood samples were detected, 25 (13.02%) for TS combine β+/βN; 57 (29.69%) for TS combine β0/βN; 26 (13.54%) for TT combine β+/βN and 84 (43.75%) for TT combine β0/βN. Among 142 prenatal samples, there were 7 (4.93%) TS combine β+/βN; 39 (27.47%) TS combine β0/βN; 21 (14.79%) TT combine β+/βN; 64 (45.07%) TT combine β0/βN; 6 (4.23%) Hb H combine β-thalassemia carriers; 2 (1.41%) Hb Bart’s combine β-thalassemia carriers and 3 (2.11%) TM/TI combine α-thalassemia carriers.
Genotypes and Mutation spectrum Identification
In all, we detected 35 mutation types of α-thalassemia, including 12 deletions, 15 non-deletion and 8 co-inheritances of deletion and non-deletion mutations. --SEA/αα, -α3.7/αα and -α4.2/αα were top 3 mutations in peripheral blood samples, while in prenatal samples they were --SEA/αα, --SEA/--SEA and -α3.7/αα. For β-thalassemia, a total of 50 (48 non-deletion and 2 deletion) types of mutation were detected, and the most common mutations in both prenatal and peripheral blood populations were βCD41−42/βN, βIVS−II−654/βN and β−28/βN. For co-inheritance of α- and β-thalassemia, 42 types of mutation has been detected, in peripheral blood samples, the top 3 mutations were --SEA/αα combine βCD41−42/βN, --SEA/αα combine βIVS−II−654/βN and -α3.7/αα combine βCD41−42/βN; while in prenatal samples, they were --SEA/αα combine βCD41−42/βN, --SEA/αα combine βIVS−II−654/βN and -α3.7/αα combine βIVS−II−654/βN or --SEA/αα combine β−28/βN. Besides, we also tested 26 rare mutations and discovered 5 novel mutations that have never been reported in HbVar, 14 ITHANET 15 database or paper. The five mutations included NC_000016.9:g.223681-227492del3812; HBA1:c.301-31_301-24delCTCGGCCCinsG and HBA2:c.95+7C>T for α-thalassemia, HBB: c.315+143G>A and HBB:c.263_276delCACTGAGTGAGCTG for β-thalassemia. All the novel mutations were verified by sanger sequencing (Figure 2). The detailed mutation types of thalassemia were listed in Table 4.

Sanger sequencing verification results of 5 novel mutations. A: NC_000016.9:g.223681-227492del3812; B: HBA1:c.301-31_301-24delCTCGGCCCinsG; C: HBA2:c.95+7C>T; D: HBB: c.315+143G>A; E: HBB:c.263_276delCACTGAGTGAGCTG. The arrows indicate breakpoints/mutation position.
Mutation Spectrum of Thalassemia
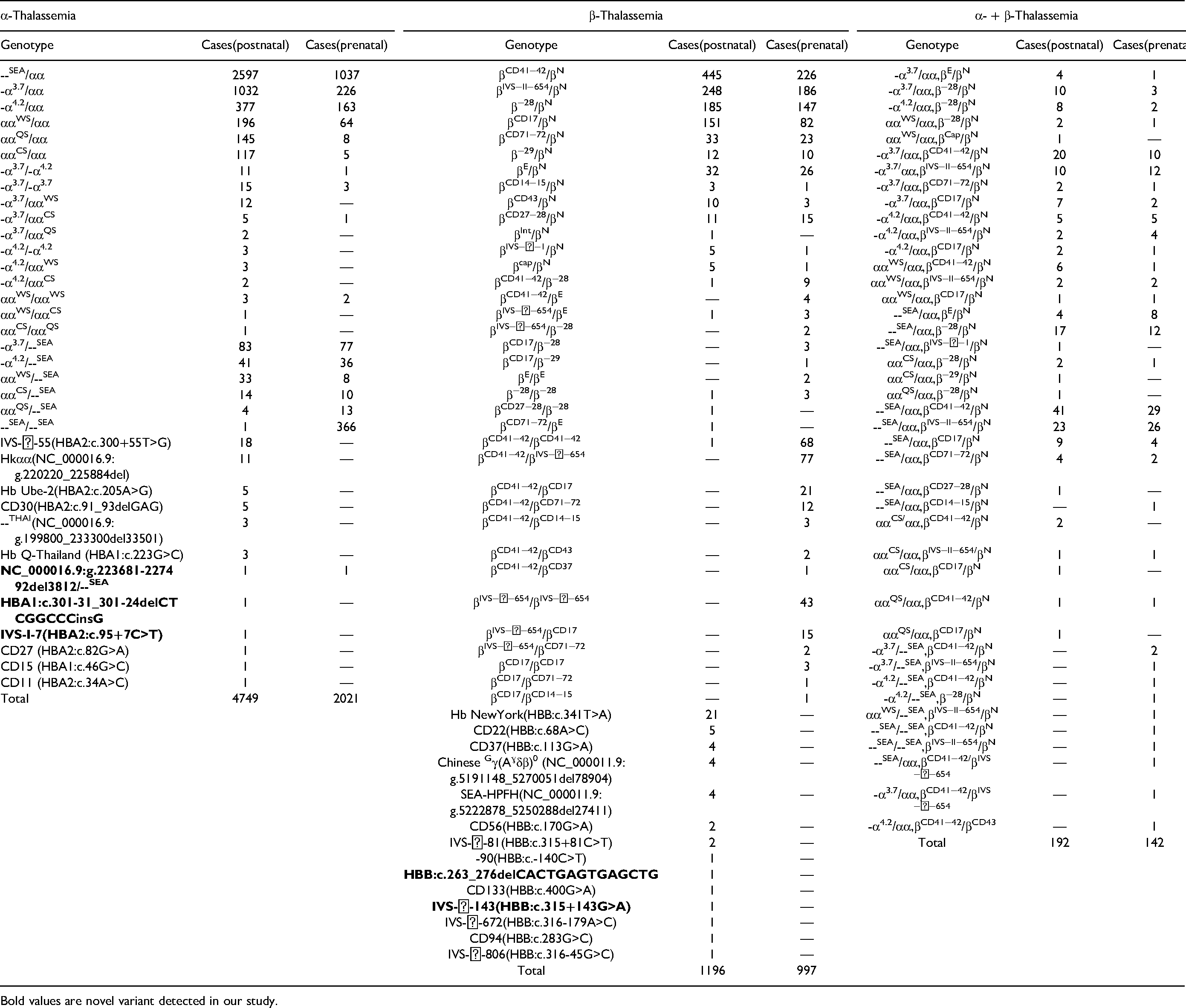
Bold values are novel variant detected in our study.
For the 3.812kb deletion, special primers F (5′-CTCGGTAGCCGTTCCTCCTGC-3′) and R (5′-CTCCTCCCGCCCCTGCCTTTTC-3′) were designed for the identification. The control and father samples had a band of 4042bp while the mother and fetal samples only showed a band of 230bp through gel electrophoresis analysis.
Discussion
In Guangdong, thalassemia is a high-incidence genetic hemolytic disease, which poses a serious threat to human life and health. Currently, the generally used effective method to prevent the birth of children with thalassemia is gene screening combined with prenatal diagnosis for pregnant-age couples.16–18 According to the analysis in the peripheral blood samples from 31 512 couples/probands and 3828 prenatal samples, and retrospective analysis of genetic testing, our study found that the carrying rate of α-thalassemia was higher than that of β-thalassemia. For mutation types, α-thalassemia was mainly large deletion type, while β-thalassemia was mainly point mutation, which is consistent with Guangdong and Guangxi, the proportion of mutation types is also in line with the mutation frequency characteristics of the southern Chinese population. α-thalassemia mainly had --SEA/αα, -α3.7/αα and -α4.2/αα deletion types, and high incidence of β-thalassemia in our study were βCD41−42/βN, βIVS−II−654/βN, β−28/βN and βCD17/βN, which is consistent with Zhao’s research in 2020. 19 The Chinese Gγ (Aγδβ)0 and SEA-HPFH were the most common large deletion among them. 20 The rare types of α-thalassemia were IVS-II-55 and HKαα, while the highly prevalent of β-thalassemia were Hb New York and CD22. All the rare types of thalassemia mutation participants showed a minor phenotype. Interestingly, we found that all the Hb New York present hemoglobin K in capillary electrophoresis results, so we supposed that it can be directly identified by detecting hemoglobin Z11 (hemoglobin K).
In this study, we characterized the novel -α3.812/--SEA deletion using specific primers for the first time. For the family, common Gap-PCR of α-thalassemia deletion showed the mother and fetal were --SEA/αα, but the RDB for α-thalassemia non-deletion showed blank for both wild and mutation. Comprehensive analysis suggested that the sample had a rare type of α deletion, which leads to the failure of non-deletion α amplification. The 3.812kb deletion started from 3′UTR of HBA2 gene to 3′UTR of HBA1 gene, but the phenotype of the mother (HGB:82g/L, MCV:65.7fL, MCH:19.5pg) was only thalassemia trait, and the father’s genotype was --SEA/βCD41−42, so they decided to give birth to the fetal. We will continue to follow up and improve the clinical data for this family. The hematological data of the remaining 4 novel mutations were HBA1:c.301-31_301-24delCTCGGCCCinsG (HGB:103g/L, MCV:67fL, MCH:19.4pg, Hb A2:2.1); HBA2:c.95+7C>T(HGB:105g/L, MCV:72.5fL, MCH:22pg, Hb A2:2.2); HBB:c.263_276delCACTGAGTGAGCTG(HGB:78g/L, MCV:73.8fL, MCH:22.5pg, Hb A2:4.7); HBB: c.315+143G>A(HGB:139g/L, MCV:90.6fL, MCH:31.9pg, Hb A2:2.7, Hb F:2.8), respectively. All of them were thalassemia trait except for the HBB: c.315+143G>A variant, which only showed thalassemia silent because of increased Hb F level. The regulation mechanism is unknown, it may be associated with some modified genes such as KLF1, BCL11A, HBS1L and MYB.21–23 We need more testing of modified gene mutations to explore its mechanism of regulating clinical phenotypes. In order to prevent and control thalassemia, different detection methods should be adopted for different mutation types.
The hematological data for each type of thalassemia are consistent with previous data, for common α-thalassemia, only --SEA/αα had a significant difference in HGB, MCV, MCH and Hb A2 level; -α3.7/αα, -α4.2/αα and ααWS/αα had a significant difference in MCV level; αCS/αα seems to be more severe cause their Hb A2 also had a significant lower level, while αQS/αα seems to be the mildest phenotype with only MCV levels decrease. When do thalassemia screening, the MCV and MCH levels of 3.7, 4.2, WS and CS types could also larger than 80fL and 27pg. This reminds us that in clinical work, we should not only look at the abnormality of these two indicators to detect thalassemia genotype. In Figure 1B, it is shown that TS (36.26%) and TT (60.03%) cases which is almost 96% of the α-thalassemia population are almost not showing any significant hemoglobin, MCV, MCH and Hb A2 differences in comparison to healthy controls, so most of them do not know that they are thalassemia carriers and their normal lives will not be affected, routine physical examination is difficult to find them, unless a specific thalassemia gene experiment. But if both husband and wife are thalassemia gene carriers, there is a 25% chance of giving birth to a child with thalassemia intermedia or major, and 50% chance of giving birth to a carrier. So It’s very important for the government to lead pre-pregnancy screening to reduces birth defects in areas with high incidence of thalassemia. The distribution of α+ and α0 thalassemia in Africa, Mediterranean, Middle East, South Asia, East Asia and South East Asia are 5-40% (-α3.7,αTα), 5-15% (--MED,-α3.7,αTα), 60% (-α3.7,αTα), 15-80% (-α3.7,-α4.2), 5-15% (--SEA, -α3.7,-α4.2,αTα), 5-80% (-α3.7,-α4.2,αTα) respectly, while the --SEA genotype mainly exists in some East Asia and South East Asia countries. 24 As a referral center, our screening for carrier rates are higher than general population screening, so the 96% only represents the distribution of mutation types rather than the prevalence, In 2007, Sorour et al reported the α-TS and TT accounts for 84.62% of α-thalassemia in multiethnic population 25 (including Pakistani, Indian, Bangladeshi, Afro-Caribbean, Yemeni, South East Asian and White Northern European). Gilad et al also found the -α3.7 and --MED genotype accounts for 70.6% of α-thalassemia deletion type in 895 mutated alleles in Jewish, ArabMuslim and South East Asia populations. 26 The composition ratio of α-TS and TT has been reported as 86.71% by Xuan et al in 2017, 27 some similar studies also showned the composition ratio of α-TS and TT data in Baoan 28 and Dongguan 29 region are 96.47% and 96.90% respectly. Actually, Guangdong has included "blood routine and hemoglobin electrophoresis" of thalassemia screening project into the government-subsidized routine obstetric examination for pregnant women. In our local practice, when women were screened positive for α-thalassemia at our hospital, we usually invite their husband to be screened instead of performing a genetic diagnosis immediately. If both couples were positive for α-thalassemia trait, then a prenatal diagnosis is required to prevent of having a hydrops fetalis.
For β-thalassemia hematological data, all mutation types showed decreased MCV and MCH levels and increased Hb A2, the latter one is being a well-known useful parameter for distinguishing thalassemia types. For co-inheritance of common α- and β-thalassemia (Table 1), the Hb A2 level of all thalassemia groups increased significantly, but their HBG, MCV and MCH levels can be in a normal range, this phenomenon needs to be distinguished from β-thalassemia. The hematological and genotype data of β-thalassemia intermedia and major patients is listed in Table 3, There are only 6 patients of β-TI/TM, the βCD27−28/β−28 and βCD71−72/βE genotypes are pregnant women in high-risk obstetrics, the remaining 4 cases are postpartum children/probands. Hematological parameters are not available for intermedia and major prenatal samples due to labor induction.
The 2004 epidemiological survey of thalassemia in Guangdong showed that the total carrier rate was 11.07%. In 2017, the Guangdong Provincial Office of Thalassemia Prevention and Control reported that the thalassemia carrier rate was 16.83%, while the carrier rate obtained from our study was 19.48%, which was higher than the previous two epidemiological surveys. One of the possible reasons is that the prenatal diagnosis department has referral cases with screening positive patients rather than the results of the extensive gestational age screening; the second reason is that our study improved the clinical pathway of conventional thalassemia screening, so that more previous missed cases and rare thalassemia mutation types have emerged. Our results also show the characteristics of thalassemia in Guangdong are mainly composed of α-thalassemia (15.07%) followed by β-thalassemia (3.80%) and co-inheritance of α- and β-thalassemia (0.61%) cases. The prevalence of rare type α- and β-thalassemia are 0.77% and 2.23% respectively.
For the prenatal cases, as high as 82.55% was diagnosed as thalassemia also demonstrated that our prenatal diagnosis has effectively prevented the birth of moderate to severe thalassemia children and improved the quality of the population. The genotype profile of the parents tested for prenatal diagnosis are listed in Supplemental Table 1, there are 5750 α-thalassemia and 1502 β-thalassemia and 404 co-inheritance, majority of them are TS or TT. Usually there are three situations in which prenatal diagnosis is performed for the fetus: 1. Both couples undergo hematological examinations for thalassemia screening in our hospital. If at least one has a positive hematological (MCV<82fL, MCH<27pg, HBA2>3.5% or <2.5%), further genetic diagnosis will be carried out for them. If it is possible to give birth to children with thalassemia intermedia (TI) and thalassemia major (TM), prenatal diagnosis will be performed; 2. Both couples had screened for thalassemia in the primary hospital with positive hematology, then come to our hospital for genetic diagnosis; 3. Couples who comes with both thalassemia gene reports and may have possibility of giving birth to TI or TM children. In fact, it can be avoided by assisted reproduction, but in this study the IVF and PGT samples were excluded. The choice of assisted reproduction will also be affected by many factors such as the economic situation and the patient's education level.
When encountering rare thalassemia mutations in clinical applications, more methods are needed, such as Sanger sequencing of the entire globin gene, MLPA, specific primer Gap-PCR, and even next-generation sequencing (NGS), to verify the precise breakpoint.27,30,31 In our study, two different company kits, Yaneng and Yishengtang, were used to detect conventional thalassemia, and there was no difference in the results, indicating that both of them can meet the needs of clinical practice. It was found that the hematology results of some patients with thalassemia silent had no different from those of normal people, so traditional testing schemes are likely to cause missed diagnoses. Our research uses the traditional procedures by carrying out the thalassemia screening and routine genetic diagnosis at the same time, this can discovery more thalassemia silent, it will be great a help for those who need to do prenatal diagnosis in the future. Besides, a rare type of thalassemia exploration was arranged when clinical abnormal thalassemia patients haven’t been detected by conventional methods. This allows more thalassemia carriers with false-negative screening to be tested and provides a more comprehensive assessment of the genetic testing of thalassemia in Guangdong. It also gives clinicians accurate and timely help in prenatal diagnosis or reproductive genetic consultation.
Our research can precisely and efficiently estimate the thalassemia genotypes, and can also find rare disease-causing mutations. In the follow-up evaluation of rare type samples, serum iron data should be added before the test for more precise screening. More samples need to be enrolled in the study and further investigation with regulating genes/regions related to hemoglobin expression should be conducted.
Supplemental Material
sj-xlsx-1-cat-10.1177_10760296221119807 - Supplemental material for Molecular Epidemiology and Hematologic Characterization of Thalassemia in Guangdong Province, Southern China
Supplemental material, sj-xlsx-1-cat-10.1177_10760296221119807 for Molecular Epidemiology and Hematologic Characterization of Thalassemia in Guangdong Province, Southern China by Jiajia Xian, Yanchao Wang, Jianchun He, Shaoying Li, Wenzhi He, Xiaoyan Ma and Qing Li in Clinical and Applied Thrombosis/Hemostasis
Footnotes
Acknowledgements
We thank all the participants for their cooperation during this work. This study was supported by the Project of the Department of Science and Technology of Guangzhou (201902010005).
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the the Project of the Department of Science and Technology of Guangzhou (grant number 201902010005).
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.