
Abstract
We analyzed data for women who received fondaparinux for ≥7 days during pregnancy. The study retrospectively included women who received fondaparinux pre-, peri- and/or postpartum for ≥7 days for prophylaxis/venous thromboembolism (VTE) treatment at German specialist centers (2004-2010). Data on pregnancy, VTE risk factors, anticoagulant treatment, pregnancy outcome and adverse events were extracted from medical records. 120 women (mean age 31.5 years) were included. Among 84 women with prior pregnancies, 41.0% had ≥1 abortion. Anticoagulation was indicated for prophylaxis in 92.5% cases, including 82.5% women with an elevated VTE risk (82.8% thrombophilia, 34.2% VTE history). All women received low-molecular-weight heparin (LMWH) as first-line therapy; 3 also unfractionated heparin. Treatment changed to fondaparinux, due to heparin allergy (41.7%) or heparin-induced thrombocytopenia (10.0%). Fondaparinux was generally well tolerated. Adverse events included bleeding events (n = 5), abortion (n = 2), premature births (n = 2), stillbirth (n = 1), arrested labors (n = 2), injection site erythema (n = 4) and unspecified drug hypersensitivity (n = 6). No VTE events or increased liver enzymes occurred during treatment. In this retrospective study, fondaparinux was effective and well tolerated. Trial registration: ClinicalTrials.gov NCT01004939.
Introduction
Women are at an increased risk of developing venous thromboembolism (VTE) during pregnancy, even with uncomplicated pregnancies. 1,2 Reasons include hypercoagulability caused by increased levels of coagulation factors and inhibitors of fibrinolysis and decreased levels of protein S, 3 compression of veins by the growing uterus resulting in impaired blood flow and venous stasis in the lower limbs, and vascular injuries during delivery. 4
Low-molecular-weight heparins (LMWH) are recommended as the preferred agents for the prevention and treatment of VTE during pregnancy. 5 - 9 LMWH do not cross the placenta, as determined by measurements of anti-factor Xa activity in fetal blood, 10 and there is no evidence that LMWH are teratogenic or increase the risk of fetal bleeding. 11,12 In pregnant women, the incidence of bleeding associated with LMWH is low, 11,13 and LMWH appear to carry a lower risk of heparin-induced thrombocytopenia (HIT), osteoporosis, and allergic skin reactions compared to unfractionated heparin (UFH). 8,14 In addition, LMWH is more convenient to use as they are usually administered only once daily, and aPTT monitoring is not required. 5,9 However, in a considerable proportion of pregnancies, heparin intolerance (allergic reactions or HIT) makes it necessary to change to an alternative anticoagulant. 15 - 17
Fondaparinux is a synthetic selective indirect inhibitor of activated factor X, whose favorable efficacy and safety profile has been demonstrated in clinical studies in various indications. 18 - 20 Fondaparinux has a once-daily dosing schedule, a low potential for causing intolerance, 21 and little or no risk of causing HIT. 22 Furthermore, no dose adjustment for bodyweight is required during prophylaxis, and platelet monitoring is not needed. There is evidence that fondaparinux passes the placental barrier to a small extent 23,24 ; however, this is considered unlikely to be of clinical significance. 24 Based largely on case reports and case series, fondaparinux has been reported to be effective and safe for VTE prophylaxis during pregnancies in which cutaneous allergic reactions to heparins or heparinoids 23 - 29 or HIT 26,30,31 had occurred and the use of an alternative anticoagulant was required. A review of 68 reported cases of fondaparinux use in pregnancy concluded that it was an effective option for preventing VTE during pregnancy in patients with HIT or allergic reactions to heparin. 32
Additional data on the use of fondaparinux for the management of VTE in pregnancy would be helpful to expand the body of evidence. The objective of this retrospective study was to systematically gather data on how fondaparinux is used pre-, peri- and/or postpartum for both prophylaxis and therapy of VTE, in order to fill a gap in the data available concerning the use of fondaparinux during pregnancy.
Methods
This retrospective, multicenter, cohort study was performed in Germany. Data were collected for women who had, during a pregnancy, been treated with fondaparinux at 1 of 7 specialist centers between 2004 and 2010. Women who were identified as qualifying for inclusion in the study were contacted and provided comprehensive information about the purpose and nature of the study, including the collection and processing of the data. For women who subsequently provided written informed consent for study participation, data were extracted from medical records and birth-related records (so-called “Mutterpass”; including fetal outcome) and entered onto standardized hard-copy case report forms. Information was only requested directly from a participant if it was not contained in the medical records. All data were retrospectively documented and in an anonymized manner, and no additional data were collected for the purpose of this study. The protocol was submitted to the responsible Ethics Committees for approval. The trial was registered as NCT01004939 at ClinicalTrials.gov.
Patient Population
The study included women who had received fondaparinux pre-, peri- and/or postpartum for at least 7 days for VTE prophylaxis or therapy. Since fondaparinux has no registered indication for this purpose in pregnancy, it was expected that participants would generally be women facing problems with LMWH/UFH, namely those with intolerance to heparins or heparinoids or with heparin induced thrombocytopenia (HIT), and/or a history of abortion, VTE, severe fetal or maternal complications during pregnancy, severe inherited or acquired thrombophilia, or long-term anticoagulation.
Data Collection
Data were collected using a paper-based case report form with the following variable domains: risk factors for thromboembolism, indication for anticoagulation (prophylaxis, treatment), antithrombotic treatment prior to fondaparinux initiation (low molecular weight heparins, unfractionated heparin, and others), thrombocyte count prior to fondaparinux initiation, fondaparinux initiation (dose), hospitalization, pregnancy related parameters and complications.
Statistical Analysis
Data summarized for this study included demographics, previous pregnancies, VTE risk factors and history of VTE events, pre-treatments, dose and duration of fondaparinux administration, maternal and fetal outcomes, complications associated with anticoagulation, and safety parameters.
As appropriate for the study design, only descriptive analyses were performed. For continuous data mean, standard deviation (SD), median, minimum, and maximum were calculated. Categorical data, including categories of continuous data, were presented in frequency tables. The statistical analysis was performed using SAS®, version 9.1 (SAS Institute Inc., Cary, NC, USA).
Results
Demography and Pregnancy History
A total of 120 women from 7 study centers were included in the analysis (Table 1). The mean age of participants was 31.5 years and 84 (70%) had been pregnant before. Of these, 48 (41.0% of the study population) had suffered from at least 1 abortion. The current pregnancy was conceived naturally in most cases (89.2%), and 116 women were expecting 1 child (96.7%). Caesarean section was performed in 35 patients (29.2%).
Demography and Pregnancy History.a
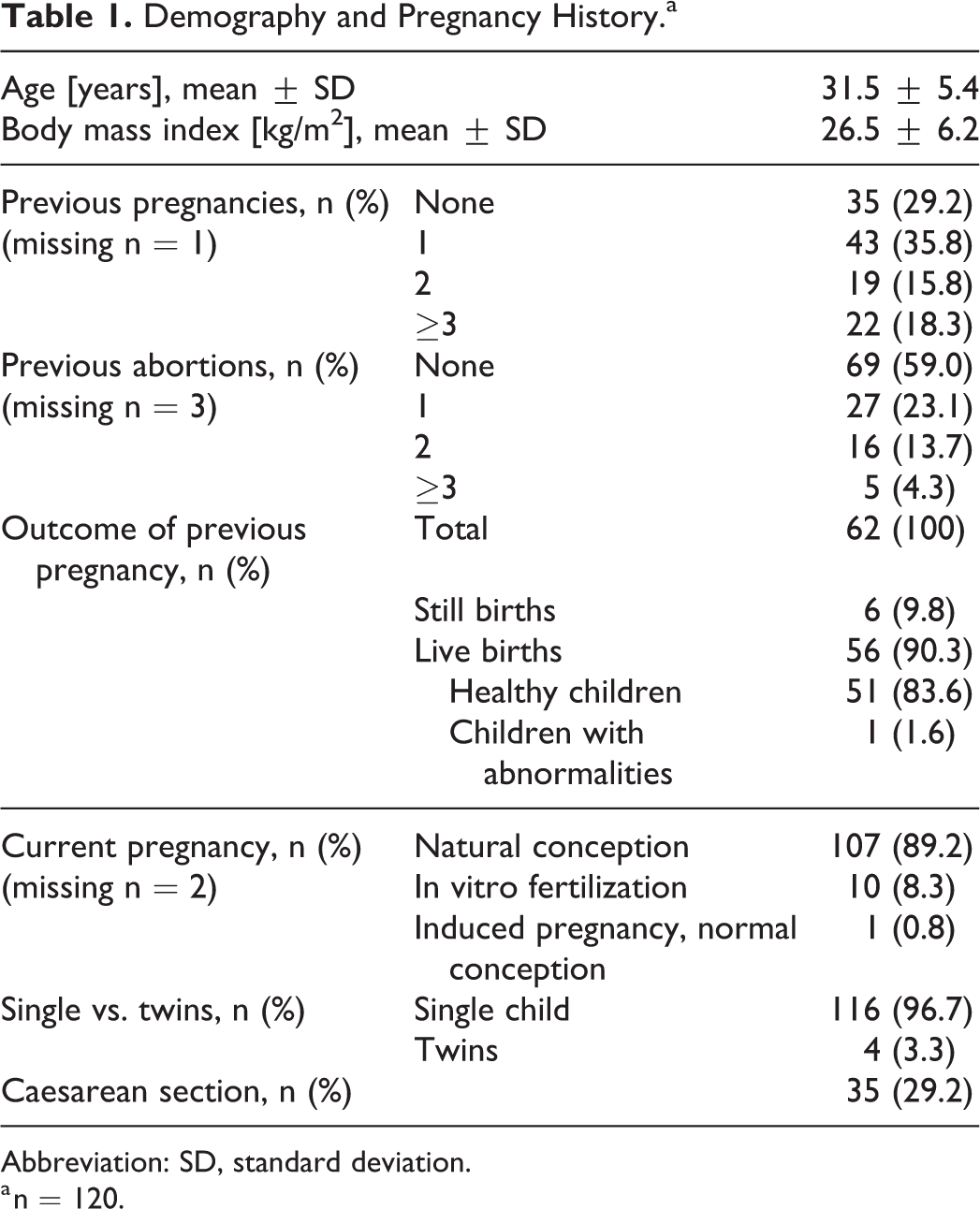
Abbreviation: SD, standard deviation.
a n = 120.
Venous Thromboembolism (VTE) Risk Factors
A total of 34.2% of participants (Table 2) had experienced thromboembolism in the past (39 women with VTE and 2 women with venous and arterial thromboembolism). In 11 patients there was a thromboembolic history of the mother, in 10 of the father, and in 11 of any sibling. The 3 most frequent thrombophilic risk factors were heterozygous factor V Leiden mutation in 35 patients (29.2%), factor XII C46T in 18 patients (15.0%), and antiphospholipid syndrome in 15 patients (12,5%). Other common risk factors were obesity (10.0%), smoking (9.2%), and previous oral contraception (9.2%).
Known Thromboembolic Risk Factors.a
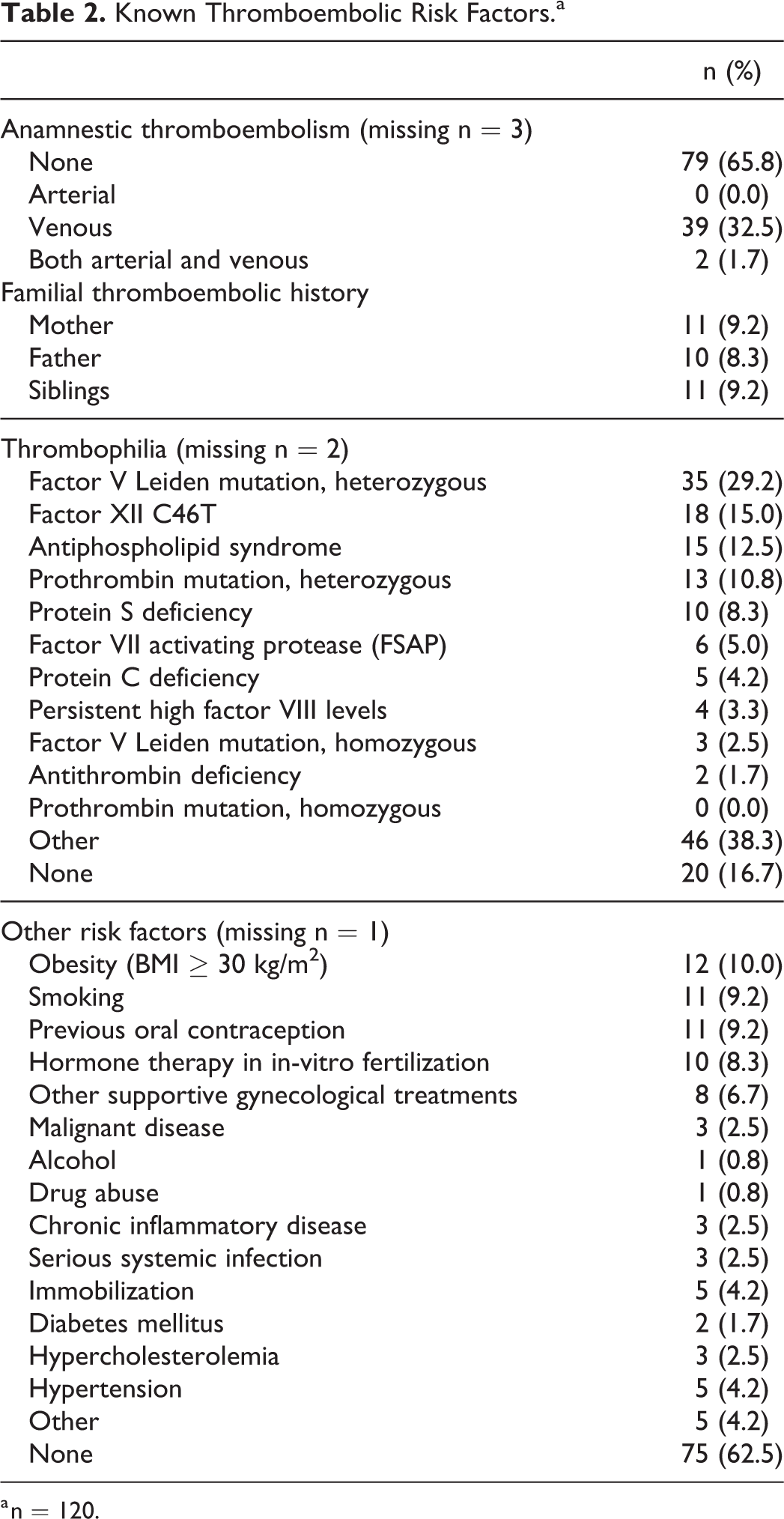
a n = 120.
Prior Anticoagulant Treatment
The reason for treatment with anticoagulants was prophylaxis of VTE in 92.5% of patients (n = 111), including 99 women (82.5%) for whom it was specifically due to an anticipated elevated risk of VTE during pregnancy. Among these 99 women, 82.8% had known thrombophilia (n = 82). Four patients had been on long-term anticoagulant therapy prior to the pregnancy, including one who had been on vitamin K antagonist treatment (phenprocoumon) because of mechanical heart valve replacement and 3 because of a history of recurrent venous thromboembolism.
All patients received LMWH prior to fondaparinux (Table 3). Three patients were initially treated with UFH and then switched to LMWH (2 because of allergic reactions to UFH). The LMWHs received included enoxaparin (23.3%), nadroparin (20.0%), dalteparin (29.2%), and certoparin (0.8%); the type of LMWH was unknown in 37.5% of patients. The duration of heparin treatment was reported for 71 patients, with a mean duration of 98.7 days.
Prior Heparin and Later Fondaparinux (Dose) Used.

Abbreviations: HIT, heparin-induced thrombocytopenia; LMWH, low-molecular-weight heparin.
a Subsequently replaced by LMWH.
The predominant reason for changing from LMWH to fondaparinux (Table 3) was the occurrence of allergic reactions to heparin (41.7% of patients). These reactions mainly involved localized erythema at injection sites, including 8 cases (6.7%) of skin necrosis; however, 12 (10.0%) women reported a generalized skin rash during heparin treatment. The time from the start of heparin treatment to the occurrence of the skin reaction was reported for 27 patients, with a mean duration of 45.7 days.
HIT during LMWH treatment was reported as the reason for changing to fondaparinux for 12 (10.0%) patients (Table 3). The mean time between the start of heparin treatment and occurrence of HIT was 157 days (median 27.5 days, range 3-576 days) among patients with available data (n = 8). An additional 4 (3.3%) patients received fondaparinux because of a drop in platelet count during heparin treatment without confirmation of HIT.
Fondaparinux Treatment
Most patients received a prophylactic dose of fondaparinux (2.5 mg daily, 78.3%) (Table 3), but therapeutic doses of up to 10 mg daily, depending upon the indication, were used in 24 patients (19.9%). The mean duration of fondaparinux treatment was 155 days, and most patients received fondaparinux after delivery as well as before delivery (Table 4). Fondaparinux treatment was interrupted for delivery, with the last pre-partum dose administered at a mean 34.6 hours before delivery, and treatment restarted a mean 11.3 hours after delivery. The most common reason for termination of fondaparinux treatment was that there was no further need for VTE prophylaxis or treatment (78.3%). A small proportion of patients were changed to another anticoagulant drug.
Characteristics of Fondaparinux Treatment.

Abbreviations: SD, standard deviation; LMWH, low-molecular-weight heparin.
Adverse Events
Seven bleeding events in 5 patients (4.2%) were reported during fondaparinux treatment, including vaginal hemorrhage in 4 patients, and uterine hematoma, abdominal wall hematoma, and hematoma in one patient each.
One patient was recorded in the case report forms as having experienced HIT while on fondaparinux. However, the corresponding investigator affirmed that there had been no case of HIT associated with fondaparinux in her cohort, but several switches to fondaparinux because of suspected HIT on LMWH had been made.
Other adverse events reported included premature birth (2 patients, 1.7%), stillbirth (1 patient, 0.8%), abortion (1 patient, 0.8%), induced abortion (1 patient, 0.8%), arrested labor (2 patients, 1.7%), and drug hypersensitivity (unspecified type in 6 patients [5.0%], injection site erythema in 4 patients [3.3%]). One patient complained of generalized itching that was already present during heparin treatment. Five (4.2%) women were hospitalized during fondaparinux treatment. No cases of VTE or thrombophlebitis occurred during fondaparinux treatment.
Outcome of Pregnancy
For 59.2% of women (n = 71), the delivery was spontaneous (Table 5), whereas 28.3% had a caesarean section, 2.5% had induced labor and 0.8% both. In total, 120 infants were born (124 fetuses including 4 twins at pregnancy start; subsequently, 2 spontaneous abortions, 1 induced abortion, and 1 stillbirth occurred). Two infants were preterm deliveries, 1 infant had thrombocytopenia at birth (the mother had immune thrombocytopenia), and 1 had petechiae on the head which resolved after 2-3 days. No malformations were reported. Most infants (75.0%) were breastfed while their mother was receiving fondaparinux treatment (data missing for 19 [15.8%]).
Outcomes.

Discussion
Alternatives to conventional UFH and LMWH are needed for pregnant women with heparin intolerance who require anticoagulant treatment or prophylaxis of VTE. Typical reasons for changing from heparins to alternatives such as fondaparinux include allergic reactions at injection sites, generalized rashes, HIT, and increased liver enzymes related to heparin treatment. 9,14,17,33 - 35 Direct oral anticoagulants rivaroxaban and edoxaban are contraindicated during pregnancy. Apixaban and dabigatran have not been associated with embryotoxicity so far, but close surveillance is recommended in these patients; vitamin K antagonists should be avoided during pregnancy because they cross the placental barrier. 7 - 9,36 Consequently, other than danaparoid (an indirect factor Xa inhibitor), there are few alternatives to fondaparinux in cases of heparin intolerance. 36,37
The current study involves the largest case series of pregnant women who received fondaparinux published so far. The majority of the 120 women included in the study received prophylactic-dose fondaparinux, although higher doses were used in some patients.
Fondaparinux was given for a mean of 155 days (range 2-1,392), with administration interrupted before delivery and restarted approximately 11 hours after delivery. The terminal half-life of fondaparinux is reported to be 17.2 ± 3.2 hours, 38 with the plasma concentration remaining above half-maximal level for 11 ± 1.4 hours. Therefore, pausing fondaparinux for 24-48 hours prior to delivery seems reasonable. Our study found that the last prepartum dose of fondaparinux was given a mean of 34.6 hours before delivery (range 12-48 hours), and this was not associated with an unexpected rate of bleeding. An alternative approach for pregnant patients receiving fondaparinux, proposed by Elsaigh et al, 39 is to switch to LMWH 1 week prior to delivery, based on an anticipated higher bleeding risk associated with fondaparinux. In our view, this is not feasible for most patients, because the reason for changing to fondaparinux is usually heparin intolerance.
The data obtained on pregnancy outcomes and adverse events in our study did not indicate any unexpected risks. Fondaparinux was well tolerated by most patients with allergic reactions to heparins, as well as by patients with previous HIT or acute HIT, or with elevated liver enzymes in response to heparin. Adverse events, such as bleeding, preterm delivery, or unsuccessful pregnancy outcome, were within the expected rates for the population and confirm the previous data on fondaparinux use in pregnancy, which was reviewed by De Carolis et al. 32
Fondaparinux does not appear to cross the placental barrier to a significant extent in experimental settings, 40,41 and only minimal concentrations of fondaparinux were found in umbilical cord blood drawn after delivery in women receiving fondaparinux, with levels at the lower margin of the measuring range of the assay 42,43 and well below the range observed in patients receiving fondaparinux for prophylaxis of VTE in orthopedic surgery. 42 Consequently, no relevant anticoagulant effect of fondaparinux is expected in infants. Furthermore, fondaparinux is specifically bound to antithrombin in human plasma, with no other relevant interactions. 44 In particular, it has minimal interaction with platelet factor 4 (PF4); this means it is unlikely to induce HIT, because the latter is caused by antibodies against multimolecular complexes of PF4 and heparin. 16,45
One of the advantages of fondaparinux is its dosing schedule. Conventional LMWHs, such as dalteparin, have a short half-life, with values between 3.87 ± 1.15 hours and 4.92 ± 2.80 hours reported in a study by Blombäck et al. 46 Thus, a once-daily dose of LMWH appears insufficient to provide a continuous protective effect against thromboembolic events for 24 hours. In contrast, a once-daily dose of fondaparinux results in a steady anticoagulant effect. However, this does mean that the interval between the last pre-partum dose of fondaparinux and delivery must be extended compared to conventional LMWH (twice the half-life).
One final observation of interest from the study concerns the temporal pattern for the development of HIT. Clinical manifestations of HIT are usually expected to develop within 5-14 days after initiation of heparin. 16 In the current study, among the subgroup that had developed HIT while receiving heparin, the occurrence of HIT was not limited to the initial phase of heparin treatment, but was reported to develop after a median of 27.5 days of treatment (range 3-576 days). While it would appear that a disproportionately large number of patients enrolled in our study had had allergic reactions to LMWH, these patients were from a pre-selected patient population and are not representative of women receiving LMWH in Germany. The incidence of HIT with LMWH is <1%, with being female, being a post-operative patient, receiving prophylactic doses of heparin and having the Tyrosinkinase JAK-2 V617F mutation increasing the risk of developing HIT. 47 - 49 Two of these factors might explain the higher incidence of HIT in our pre-selected population, but this does warrant further investigation.
When generalizing the observations made in this study, some limitations arising from the retrospective design should be taken into consideration. According to applicable international guidelines, anticoagulation is not indicated in a notable proportion of pregnant women. However, the retrospective nature of this study means that the decision to initiate such treatment was at the discretion of the prescribing physician. Data quality very much depends on the availability and accuracy of medical records. In our study information had to be gathered from different sources, i.e. the mother’s maternity records (“Mutterpass”) and the records of maternity units from the same or from different hospitals. There was no control of pre-medications, or the duration of fondaparinux exposure or the doses used, and the women were all sequentially exposed to both LMWH and fondaparinux; therefore, direct comparisons between these treatments should be interpreted with caution and no firm conclusions can be drawn concerning the efficacy and safety of fondaparinux as an anticoagulant during pregnancy. We did not collect any data concerning peridural anesthesia which would be worth another investigation in the context of fondaparinux use during/after pregnancy. An additional factor was that one center contributed a large amount of data (n = 74 women, 62%). However, sensitivity analyses performed to evaluate a possible resulting bias did not indicate relevant differences between the results of this one center compared to the results of the remaining 6 centers. On the other hand, strengths of the study included the systematic approach to identifying all pregnancies in which fondaparinux was used for 7 or more days, and the methodical effects made to capture all relevant outcomes.
Conclusions
The results of this retrospective study indicate that fondaparinux could be an effective and well-tolerated alternative for prophylaxis or treatment of VTE in pregnant women especially in cases with intolerance to LMWH.
Footnotes
Authors’ Note
CED, JK, ELL, IBG, GK, US, SK, and AH conceived, designed and conducted the study. CED, JK, ELL, BL, IBG, GK, US, SK, PB and AH interpreted the data. SE, CED, PB, and AH performed the explorative analysis and wrote the first draft of the manuscript. JK, ELL, BL, IBG, GK, US, and SK revised the manuscript for important intellectual content. All authors approved the final version of the manuscript. Data is available upon reasonable request from the author. The protocol was approved by the local ethics committees and is listed on ClinicalTrials.gov (NCT01004939). Written informed consent from patients was obtained prior to searching medical records.
Acknowledgment
Dr. Hartmut Pollmann participated in the study as investigator and in the development of this manuscript; however, he passed away before the final draft was completed and the authors would like to acknowledge his contributions.
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: CED discloses speaker honoraria from Aspen, Sanofi-Aventis, Mitsubishi Pharma, ITF Pharma, Bayer Vital Pharma, Bayer Healthcare, Daiichi Pharma, CSL Behring, Pfizer, BMS. JK discloses speaker honoraria from Aspen, Bayer Health Care Pharmaceuticals, Daiichi Sankyo, Boehringer Ingelheim, CSL Behring, Sanofi-Aventis, Pfizer, BMS, Mitsubishi Pharma, Ferring GmbH, Mylan Healthcare GmbH and Novo Nordisk. JK is also a medical advisor for CSL Behring International, Bayer HealthCare Pharmaceuticals (national and international) and Novo Nordisk (national) during the last 3 years. IBG, GK, and BL declare to have no conflicts of interest. ELL discloses speaker honoraria from Aspen, Mitsubishi Pharma, Bayer Healthcare, Daiichi Pharma, CSL Behring, Pfizer, BMS. US discloses speaker honoraria from Bayer Healthcare, CSL Behring, Pfizer, Roche. SK was previously employed by GlaxoSmithKline and owns GSK stock. SE was previously employed by GlaxoSmithKline and owns/owned no GSK stock. PB reports to have received consultancy honoraria from Aspen. AH was previously employed by GlaxoSmithKline and owns/owned no GSK stock. He is now employee of Aspen.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: GSK provided financial support for the conduct of this study and developed the study design together with the authors. The data management, analysis and report preparation were performed by the CRO Institut Dr. Schauerte, Munich, Germany. The interpretation of data and decision to submit the article for publication was made by the authors.