
Abstract
Since the onset of the global pandemic in early 2020, coronavirus disease 2019 (COVID-19) has posed a multitude of challenges to health care systems worldwide. In order to combat these challenges and devise appropriate therapeutic strategies, it becomes of paramount importance to elucidate the pathophysiology of this illness. Coronavirus disease 2019, caused by the novel severe acute respiratory syndrome coronavirus 2 (SARS-CoV2), is characterized by a dysregulated immune system and hypercoagulability. COVID-associated coagulopathy (CAC) was recognized based on profound
Introduction
The severe acute respiratory syndrome coronavirus 2 (SARS-CoV2) has led to a global outbreak of coronavirus disease 2019, termed COVID-19.
1
Health care workers have been posed with innumerous and unique challenges having to adapt at a rapid pace to prioritize the lives of patients affected by this potentially fatal virus. One of the challenges has been keeping up with the unfolding manifestations of the virus. There have been many published reports detailing these manifestations from China and Italy. Despite the rapid pace at which new information is being published, there remain many questions surrounding the pathogenesis and management of COVID-19. It is now widely recognized that COVID-19 is a systemic disease marked by a dysregulated immune system and a hypercoagulable state. Mortality in COVID-19 has been shown to correlate with both elevated interleukin-6 (IL-6) levels and elevated
In this review, we aim to discuss the various components of immunothrombosis, including key mediators such as leukocytes and cytokines, in rendering a procoagulant state and the ultimate result of thrombosis, as it relates to infectious and septic states. We also aim to unravel and illustrate the aspects of COVID-19-associated coagulopathy (CAC). Finally, we conceptualize the pathophysiology of a hypercoagulable state and thrombosis associated with COVID-19 relating to the well-described model of immunothrombosis. Although this is no longer a novel concept, it remains a relevant concept during the current pandemic of COVID-19.
COVID-19-Associated Coagulopathy and Cytokine-Release Syndrome
SARS-CoV2, a novel strain of the single-stranded RNA virus of the coronavirus family, has led to several systemic manifestations, most recognizably acute respiratory failure evidenced by diffuse bilateral infiltrates on chest imaging and progression to acute respiratory distress syndrome (ARDS). 4,5 The predominant underlying mechanism in COVID-19-related mortality is hypothesized to be widespread tissue damage and endothelial injury from an overactivated immune system via exaggerated T-cell responses and increased cytokine secretion, leading to a cytokine storm. 6 COVID-19 patients demonstrate markedly increased IL-6, IL-2R, IL-10, and tumor necrosis factor α (TNF-α) levels. 7,8 These patients are also reported to have strikingly elevated ferritin and C-reactive protein levels, akin to patients presenting with hemophagocytic syndrome or cytokine-release syndrome (CRS). 9 These levels appear to be directly associated with increased mortality. 9
With increasing numbers of patients with COVID-19, reports of aberrant hematologic parameters became evident, leading to the recognition of CAC. It has been reported that thrombocytopenia happens more often in patients with severe disease and potentially correlates with increased mortality. 10,11 Lymphocytopenia is also more prominent among severe cases. 10,12
Another prominent finding, which correlates with mortality, is the markedly elevated
Overview of Hemostasis
The inciting event for hemostasis occurs with disruption of the integrity of the endothelial lining (Figure 1). When vessel wall injury occurs, collagen and tissue factor (TF) contained in the subendothelium become exposed, triggering the formation of a thrombus. Platelet adhesion at the site of injury occurs via interactions of glycoprotein VI to exposed collagen and glycoprotein Ib-V-IX with collagen-bound von Willebrand factor (vWF). 17 Platelet activation is initiated following this interaction. Tissue factor–mediated platelet activation is an alternate pathway, which is independent of endothelial injury. 17,18 Platelet activation leads to the release of the contents of α granules and dense granules. Adenosine diphosphate (ADP) and calcium ions are released from dense granules, further stimulating platelet activation through 2 ADP receptors, P2Y1 and P2Y12. 19

Overview of mechanisms of hemostasis. The coagulation cascade during hemostasis is initiated with the release of tissue factor (TF). (1) At the site of endothelial injury, TF that is stored in the adventitial and medial layers of the vessel wall is released. Platelet activation and adhesion occurs at the site of injury. (1) Encrypted TF is also released from monocyte-derived microparticles. Encrypted TF is activated through the secretion of protein disulfide isomerase (PDI), which occurs because of endothelial injury and platelet activation. (2) Activated TF will form a complex with circulating factor VIIa (FVIIa), leading to activation of FX to FXa. A small amount of thrombin generated creates an amplification loop by activating FVIII, FXI, and FV. (3) During the propagation phase, FIXa, which is generated by TF–VIIa complex or FXIa, binds to its cofactor FVIIIa to form the intrinsic tenase complex. FXa is then formed by TF–FVIIa complex or FIXa–FVIIa complex. (4) FXa binds to FVa, forming the prothrombinase complex. (5) FXa–FVa complex converts prothrombin to thrombin. (6) Thrombin further activates fibrinogen to fibrin, which is cross-linked by FXIIIa to form a stable clot.
Concurrently, the coagulation cascade begins with the activation of TF, which is a potent initiator of thrombin generation and fibrin formation. Tissue factor is expressed in 2 major anatomical compartments that are involved in coagulation: the adventitia and the medial layer of the vessel wall, and microparticles circulating in the blood. 17 When the endothelial layer of the vessel is structurally damaged, TF is exposed and activated (Figure 1). Additionally, upon platelet and endothelial activation, secretion of protein disulfide isomerase (PDI), dubbed the “damage signal,” activates encrypted TF within the microparticles (Figure 1). 17 Activated TF will form a complex with Factor VIIa (FVIIa) to activate FIX, FX, and additional FVII. Generated thrombin will activate FXI, FVIII, and FV. Factor IXa, which is generated by TF–VIIa complex or FXIa, binds to its cofactor FVIIIa to form the intrinsic tenase complex, leading to activation of FX (Figure 1). The resultant FXa and FVa complex converts prothrombin to thrombin, leading to propagation of coagulation cascade (Figure 1). 17 Thrombin further activates fibrinogen to fibrin, which will be cross-linked by activated FXIIIa to form stable thrombi (Figure 1). Fibrin formation is counterbalanced by the fibrinolytic system, by tissue-type plasminogen activator (tPA), which cleaves plasminogen to form the serine protease plasmin, which degrades fibrin to limit the extent of thrombus formation. 20
Immunothrombosis
The term “immunothrombosis” was conceptualized by Engelmann and Massberg in 2013 3 to accurately define the intricate network between the coagulation system and the innate immune system. This theory defines the symbiotic relationship between these distinct pathways, specifically in the context of acute infectious states. Immunothrombosis describes the active participation of the innate immune system in forming a thrombus via distinct cellular and molecular interactions, triggered by recognition of pathogens and damaged cells. Under this theory, this response is proposed to be a conserved evolutionary defensive link to inhibit pathogen dissemination. 21,22 Although there is overlap, immunothrombosis suggests mechanistically distinct pathways of thrombosis and hemostasis through the influence of the innate immune system. 23
Although the extent of the role of microthrombi in the prevention of dissemination of pathogens in septic states has been challenged, 24 the concept of immunothrombosis, nevertheless, underscores the crosstalk between the coagulation and the immune system. Furthermore, dysregulation of immunothrombosis sets up a vicious cycle of unchecked immune activation and thrombus formation. 3,25 Below, we describe the processes involved in rendering a procoagulant state in infections.
Leukocyte-Mediated Procoagulant State
Leukocytes have recently been highlighted in thromboinflammatory diseases as vital components and propagators on thrombosis. Leukocytes, particularly monocytes and neutrophils, activate platelets and the coagulation cascade through various processes (Figures 2 and 3). 26 Also, they produce cytokines, which confer a proadhesive and procoagulant effect on endothelial cells (ECs). 27 Conversely, leukocyte chemotaxis and phagocytic functions coordinate thrombus resolution. 27 Nonetheless, imbalances in the immune system threaten these carefully regulated processes, imposing procoagulant conditions (Figures 2 and 3).

Principles of immunothrombosis. Monocytes and their microvesicles release activated intravascular tissue factor (TF) in response to pathogen-associated molecular patterns (PAMPs) or damage-associated molecular patterns (DAMPs), which initiates the extrinsic pathway of coagulation. Activated platelets and endothelium secrete protein disulfide isomerase (PDI), which activates microvesicle-derived TF. Interaction between P-selectin on activated platelets and P-selectin glycoprotein ligand (PSGL) on leukocytes enhance recruitment of leukocytes and increase TF production. Neutrophil extracellular traps (NETs) provide a scaffold consisting of DNA, histones, and neutrophil serine proteases, and they support immunothrombosis through several pathways. (1) NETs bind to von Willebrand factor (VWF) and support the recruitment of platelets. (2) Histones H3 and H4 on NETs can trigger the activation of platelets. (3) NETs bind to TF leading to TF-mediated thrombin generation through the extrinsic pathway (dotted arrow). (4) Neutrophil elastase and other neutrophil serine proteases cleave and inactivate anticoagulants, including TF pathway inhibitor (TFPI) and thrombomodulin. (5) NETs can directly activate factor XII (the contact pathway of coagulation) mediated by platelet-derived polyphosphates (PolyP). The complement system (specifically the activated complement components C3a and C5a) also enhances platelet activation.

Summary of the mechanisms of leukocyte induced procoagulant state. Activation of leukocytes during states of sepsis and inflammation leads to a prothrombotic state, primarily through platelet activation, activation of coagulation cascade, and downregulation of anticoagulant pathways.
Leukocytes lead to platelet activation
Platelet activation plays a critical inciting role in thrombosis following vascular injury. Platelets support thrombosis through the release of coagulation factors and exposing negatively charged phospholipids that act as essential cofactors for the proteolytic reactions triggered by coagulation factors. 28 Platelets and leukocytes not only engage in bidirectional pathways intended to enhance host defense and pathogen containment but also propagate a procoagulant state as a means of defense. 29 First, platelets guide the cells of the immune system to the site of the vascular injury through mediators, including various CXC-chemokine ligands, CD40 ligand, CD154, and TREM1. 30,31
Activated cells of the innate immune system and the ensuing cytokine release induce platelet activation by releasing platelet-activating factor (Figure 3). 32 Pro-inflammatory stimuli and high shear stress also promote formation of heterotypic platelet–leukocyte aggregates through the interaction of P-selectin on the surface of platelets and leukocyte surface P-selectin glycoprotein ligand (PSGL-1). 33 This interaction not only augments the recruitment of leukocytes but also leads to increased expression of TF on monocytes (Figures 2 and 3). 27,34 The platelet–leukocyte aggregates are associated with many thromboinflammatory conditions, such as inflammatory bowel disease. 27,35 Additional interactions of platelets and vWF with neutrophil extracellular traps (NETs) are pivotal mechanisms in immunothrombosis and will be described below (Figure 2).
Leukocytes enhance the activity of the coagulation system
Leukocytes- and microparticle-derived TF is thought to play a more integral role in thrombosis than in physiologic hemostatic pathways. 3 Intravascular TF is functionally inert in its natural state, specifically when expressed on hematopoietic cells. 36 Pro-inflammatory states such as infections and sepsis provide the stimulus for activation of monocyte and microparticle-derived intravascular TF release (Figure 3). Protein disulfide isomerase is an oxidoreductase secreted by activated platelets, ECs, or by damaged cells and supports the activation of leukocyte- and microvesicle-derived intravascular TF (Figure 2). 37,38 Tissue factor decryption occurs through other various mechanisms, including P-selectin and PSGL interaction, 39 and the recognition of pathogen-associated molecular proteins (PAMPs) via toll-like receptors (TLRs) on the surface of monocytes. 22 Additionally, damage-associated molecular proteins released from damaged host cells, ECs, and consequent PDI signaling lead to microvesicle-derived TF activation (Figure 2). 40 Activated monocytes and the microparticles that they release remain the largest source of intravascular TF, 41 with neutrophils and eosinophils contributing to a lesser extent. 42 –44
Neutrophils mediate a procoagulant environment primarily through the release of NETs (Figure 3). 27 Following exposure to pathogens, activated platelets, and cytokines, neutrophils undergo the formation of NETs via a process termed NETosis (Figure 2). 3,27 Neutrophil extracellular traps constitute a matrix of DNA, histones, and various neutrophil antimicrobial machinery. 29,45 Apart from providing antimicrobial defense, NETs play a fundamental role in providing a scaffold for procoagulant effectors consisting of platelets, vWF, TF, extracellular histones, high mobility group box-1, neutrophil serine proteases, and fibrinogen (Figures 2 and 3). 26,46,47
Neutrophil extracellular traps also provide a catalytic platform for proteolytic activity of neutrophil serine proteases, such as neutrophil elastase, which degrades and alters the function of anticoagulants such as thrombomodulin. 48 Histone H4 binds to prothrombin and generates thrombin by autoactivation. 49 Neutrophil elastase and cathepsin G, which are part of the antimicrobial repertoire, also proteolytically inactivate another anticoagulant, tissue factor pathway inhibitor (TFPI), the primary inhibitor of thrombin (Figures 2 and 3). 45,50 Neutrophil extracellular traps have also been shown to bind to factor XII (Hageman factor), leading to its activation to FXIIa, prompting the direct activation of the contact pathway of coagulation (Figures 2 and 3). The contact pathway is thought to have a vital role in immunothrombosis, in contrast to its minor role in physiologic hemostasis. 3,50
Thrombosis is regulated by 3 principal anticoagulants, which are antithrombin, thrombomodulin, and TFPI. 51 The procoagulant state is further propagated during infections by decreased production of anticoagulants from endothelial injury as well as inhibition of anticoagulant mechanisms via degradation by neutrophil elastase. 51 Typically, an intact EC has anticoagulant properties. 52 The release of inflammatory cytokines during infection and sepsis significantly alters the antithrombotic nature of the EC. Glycosaminoglycans act as heparin-like cofactors that facilitate binding with antithrombin, leading to the production of a potent inhibitor of thrombin (Figure 3). 53,54 Decreased synthesis of glycosaminoglycans on the surface of EC occurs in inflammatory states. 54 Endothelial cell also expresses thrombomodulin, which binds to thrombin, leading to decreased protein C activation (Figure 3). 55 Activated protein C regulates coagulation activation by proteolytic cleavage of the essential cofactors Va and VIIIa. 55 The third, major impaired anticoagulant pathway includes TFPI, the primary inhibitor of the TF–FVIIa complex. 56,57 Neutrophil elastase plays a central role in the inactivation of TFPI (Figures 2 and 3). 51
Cytokine-Mediated Procoagulant State
Cytokines and interferons play a pivotal role in inflammation. Cytokines are secreted by activated leukocytes and ECs. 58 Its relationship with the coagulation system during inflammatory states is complex and dynamic. The cytokines exert a procoagulant effect by means of increased platelet activation, release of TF from monocytes and ECs, downregulation of anticoagulant pathways, and inhibition of fibrinolysis. 59 –62 Conversely, the production of pro-inflammatory cytokines such as IL-6 and TNF-α by monocytes through protease-activated receptors (PARs) signaling is enhanced by thrombin generation. 51
Notably, there is inconclusive evidence suggestive of an association between risk of venous and arterial thrombosis and single-nucleotide polymorphism heterozygosity in inflammatory-related genes of IL-1A, IL-4, IL-6, and IL-13. 63,64 Many of the procoagulant effects of cytokines intersect with complement-mediated pathways and have a crucial role through interactions with leukocytes, platelets, and ECs. Therefore, their in-depth mechanisms are detailed in the respective sections in this review.
Viral-Mediated Procoagulant State
The procoagulant effects of bacterial endotoxins via augmentation of TF and plasminogen activator inhibitor expression are well described. 65 Nevertheless, many viral infections can cause hemorrhagic or thrombotic complications. 66 In one such model, using mice injected with lethal doses of influenza virus developed ARDS and hyperinflammatory cytokine responses, evidence of platelet activation, microvascular thrombi, and NETosis. This model demonstrated similar findings to endotoxemic models of sepsis. 67 Viral-mediated coagulation is postulated to occur in 4 main ways: endothelial disruption, leukocyte–platelet interaction, cytokine release, and release of intravascular TF.
Endothelial dysfunction and apoptosis can occur via direct viral infection or through cytokine release. Activation of TF and vWF, as well as exposure of collagen from denuded EC and cytokine release, leads to thrombus formation through platelet activation and extrinsic coagulation cascade. 68 Furthermore, viruses can directly infect the cells of the innate immune system, which can lead to immune dysregulation and potential consequential effects on the coagulation system. 67 –69 Although TLRs, especially TLR7 and platelet interaction, are described in viral infections, the role of this association in the propagation of a thrombus is unclear. 70
In conclusion, a viral-mediated coagulant state culminates in the presence of endothelial injury and dysfunction and cytokine-driven inflammatory conditions, leading to activation of TF-mediated thrombosis.
Complement-Mediated Procoagulant State
It has been reported that complement activation enhances thrombosis at different points of the coagulation process. 71 Complements can modify phospholipid membranes of cells, which is essential for TF-initiated coagulation. 71 Studies have shown that complement pathways can trigger the activation of platelets and fibrin formation (Figure 2). 72 Besides, activated complement factors can increase TF expression in various cells and modify mast cells and basophils to a prothrombotic phenotype. 72 –74 Clinically, complement-related prothrombotic state is observed in diseases such as paroxysmal nocturnal hemoglobinuria, glomerulonephritis, and vasculitis. 75
Fibrinolysis During an Infectious State
Under physiological conditions, the innate immune system also modulates fibrinolytic pathways. Leukocytes express urokinase-type plasminogen activator (uPA) and uPA receptor (uPAR), in addition to receptors for plasminogen. Plasminogen is localized to the leukocyte surface increasing the activation of tPA or uPA. 29 Although leukocytes possess the endogenous capability to enhance fibrinolysis, excessive coagulation during a state of dysregulated immune system can overwhelm the balance to favor persistent thrombus formation. 76
Microthrombosis, VTE, and Arterial Thrombosis in Infection
Microthrombosis during infections and sepsis has been extensively described in the literature, demonstrating the crosstalk between the immune system and the coagulation pathway. Microvascular thrombosis is characterized by the formation of fibrin and platelet-rich thrombus, resulting in occlusion of microvessels (arterioles, venules, capillaries). 77 The consequences can range from transient disruptions in the coagulation system to ischemia and organ dysfunction. Microvascular thrombosis likely occurs at a more frequent rate than reported due to difficulty in establishing this diagnosis. As of yet, there is not a specific laboratory surrogate for this entity.
Venous and arterial thrombosis in sepsis with resultant organ ischemia is the telltale sign of dysregulated immunothrombosis. Recent evidence suggests that aberrant immunothrombosis also contributes to thrombus formation in large vessel disease, including atherothrombosis and DVT. 51 Structural analysis of venous clots demonstrates activated leukocytes and NETs within the architecture of the thrombus, supporting the involvement of leukocyte-mediated platelet activation and NET-driven VTE. 27,29 Arterial thrombosis occurs following plaque rupture and thrombus formation.
Leukocytes can partake in atherothrombosis in inflammatory states by generating microparticle-associated TF at the site of vessel injury. 29,78 Plasma levels of TF-expressing monocytes and microparticles, leukocyte–platelet aggregates, and NET markers are elevated in conditions that predispose to cardiovascular disease. 3,29 Additionally, pro-inflammatory cytokines destabilize the atherosclerotic plaques by promoting cell apoptosis and matrix degradation. 78 Other contributing factors that lead to arterial thrombosis in inflammatory states are demand ischemia, impaired fibrinolysis, endothelial dysfunction, and cytokine-induced vasospasm. 79,80
The current COVID-19 pandemic has resurrected the concept of immunothrombosis as it is a relevant model to demonstrate the potentiating effects of the immune system and the coagulation system and the detrimental effects associated with their unrestrained activation, as evidenced by microthrombi and overt venous and arterial thrombi (Figure 4). The impact of aberrant immunothrombosis on mortality in COVID-19 is a matter of ongoing interest and debate as identifying the most significant therapeutic target or synergistic pathways may have enormous implications on outcomes.
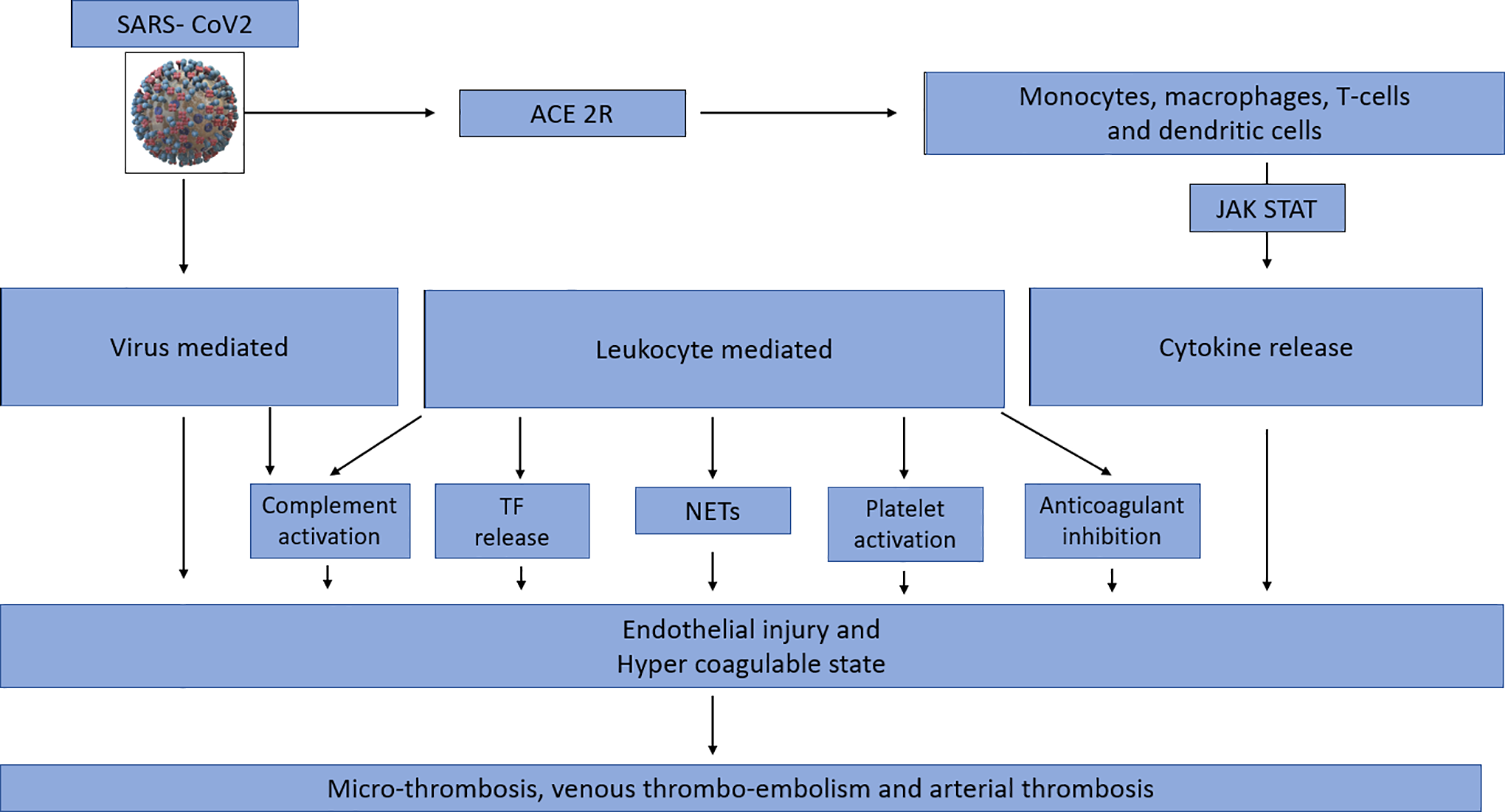
Model of COVID-associated coagulopathy. A, First, SARS-CoV2 gains entry into cells via the angiotensin-converting enzyme 2 receptor (ACE-2R), leading to activation of the innate immune system. Direct viral infection of the immune cells leads to dysregulation and cytokine release. B, Activated monocytes, macrophages stimulate Janus Kinas (JAK)- Signal transducer and activator of transcription (STAT) (JAK STAT) pathways via cis/trans signaling, leading to an amplification of cytokine release. C, SARS-CoV2 infects endothelial cells directly, resulting in endothelial injury and therefore propagating a hypercoagulable state. D, Leukocyte activation and ensuing cytokine storm in SARS-CoV2 leads to a hypercoagulable state though 5 main ways: increased tissue factor release, neutrophil extracellular traps, platelet activation, inactivation of anticoagulant pathways, and complement activation. E, Dysregulation of the immune system in SARS-CoV2 infection and the resultant endothelial injury and hypercoagulable state culminates into widespread microthrombosis, venous thromboembolism, and arterial thrombosis.
We propose the following pathways as contributing to the coagulation abnormalities and thrombosis seen in COVID-19, using supporting evidence from prior published reports.
SARS-CoV-2, a β-coronavirus, infects host cells through angiotensin-converting enzyme 2 (ACE2) receptor facilitated by the host’s TMPRSS2 membrane protease. 81 Angiotensin-converting enzyme 2 receptors are found on cardiopulmonary cells, ECs, as well as hematopoietic cells such as monocytes and macrophages. 80 The mechanisms of SARS-CoV-2-mediated procoagulant effects are described below.
Angiotensin-converting enzyme 2 has inherent anti-inflammatory properties through direct communication with macrophages and attenuation of angiotensin II levels. Under normal physiological condition, angiotensin II is pro-inflammatory. 82 Downregulation of ACE2 occurs following viral engagement, leading to elevated angiotensin II levels, further adding to a hyperinflammatory state. 83,84
Varga et al 85 recently published a case series of 3 patients with SARS-CoV2 and found to have EC infection and endothelitis. Histological examination of vascular structures of varying organs in all 3 of the patients demonstrated viral inclusion bodies, mononuclear and neutrophilic infiltration, and apoptosis of endothelial and inflammatory cells. 85 Apart from direct infection of the ECs, secondary endothelial injury also occurs as a result of widespread immune activation and cytokine release (Figure 4). 86
Interleukin-6 is suggested to be a significant contributor to the immune dysregulation and ensuing CRS that occurs in COVID-19. Interleukin-6 binds to the membrane-bound IL-6 receptor (mIL-6R) on immune cells or soluble IL-6R typically on ECs (sIL-6R), resulting in a complex with gp130, leading to downstream signal transduction via JAK-STAT pathway (Figure 4). 87
Additionally, SARS-CoV2 infection is associated with decreased circulating lymphocytes and T-cell subsets (CD4+, CD8+ T lymphocytes, and regulatory T cells) either due to selective destruction, functional exhaustion, or potentially direct viral infection. 84,88 A decreased T-cell response has injurious effects due to attenuated viral clearance and antibody production; this leads to macrophage activation syndrome and cytokine storm. 84 The end result is an amplified production of cytokines leading to microthrombosis and organ dysfunction, as evidenced by high mortality in COVID-19 patients with elevated IL levels (Figure 4).
Postmortem analysis of COVID-19 patients verifies the presence of widespread microthrombi and suggests a possible cause for mortality. Fox et al 89 described 4 cases with thrombus formation within capillaries and small vessels, surrounded by aggregates of CD4+ cells. Furthermore, the demonstration of NETs on histologic analysis supports the relevance of immunothrombosis (Figure 4). 90 Zuo et al 91 also demonstrated increased levels of cell-free DNA, myeloperoxidase-DNA, and citrullinated histone H3, which are the specific markers of NETs, in the sera of COVID-19 patients. The addition of the sera of COVID-19 patients to healthy controls further promoted NETosis. 91
Disseminated intravascular coagulopathy is the most common coagulopathy that is associated with sepsis states. COVID-associated coagulopathy differs from DIC in specific manifestations; decreased fibrinogen, significant prolongation of PT/international normalized ratio, or schistocytes are not frequently seen. A recent report by Panigada et al
92
supports the discrepancy of these claims through thromboelastography (TEG). The analysis included 24 patients who collectively demonstrated a hypercoagulable state by TEG parameters. Factor VIII and vWF levels are markedly elevated in COVID-19, reflective of endothelial dysfunction and hypercoagulability.
86,92
Elevated
Excessive complement activation likely contributes to the thrombogenic state in COVID-19 (Figure 4). Mannose-binding lectin (MBL) complement pathway binds to SARS-CoV spike glycoprotein, leading to initiation of the lectin pathways through MBL-associated serine protease-2 (MASP-2) activation. 94
Murine models have demonstrated that C3-deficient mice and C5a inhibition led to decreased localized inflammation and lung damage in SARS-CoV infection. 95 A recently published case series aimed to evaluate the contribution of the complement system in 2 subsets of COVID-19 patients with lung and skin manifestations. 96 The primary histologic findings in examined lung tissues were septal capillary injury, microthrombosis associated with extensive deposits of the terminal complement complex C5b-9, as well as C4d and MASP2. 96 In the 3 patients with retiform and purpuric skin lesions, a similar pattern of the microvascular-associated complement of deposition was noted. 96
To date, there have also been several case reports, case series, and retrospective analyses published in the literature of the thrombotic manifestations in COVID-19, including microthrombi, DVT, pulmonary embolism, myocardial infarction, and strokes. Klok et al 97 reported a 27% (95% CI: 17-37) VTE incidence and 3.7% arterial vascular events in critically ill patients with COVID-19. Tang et al 93 also reported a similar incidence of VTE.
The procoagulant pathways highlighted in immunothrombosis (Figure 4) in concert appear to overwhelm the balance of hemostasis and result in microthrombi and overt thrombus formation. Microthrombosis appears to be the more frequent finding in COVID-19, compared to VTE or arterial thrombosis. The factors that dictate the occurrence of macrothrombosis in individual patients are unclear and likely multifactorial. Factors such as genetics, underlying comorbidities, immobility, cytokine storm, and viral load are inclined to play a role in that determination.
Conclusion
COVID-associated coagulopathy exemplifies the potentiating interactions between the immune system and the coagulation pathway. A procoagulant state in COVID-19 is the result of a direct viral-related endothelial injury, leukocyte- and cytokine-mediated platelet activation, TF release, and NETosis augmented by an unchecked activation of the complement system. Aberrant activation of the immune system during SARS-CoV viral infection leads to unregulated thrombosis, presenting with widespread microthrombi, as well as manifestations of VTE and arterial events.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.