
Abstract
Current guidelines recommend low-molecular-weight heparin treatment in patients with cancer with established venous thromboembolism (VTE). The aim of this article was to study the pharmacological properties and effectiveness of tinzaparin in patients with cancer as well as its potential anticancer properties. A search of PubMed and ScienceDirect databases up to March 2016 was carried out to identify published studies that detect the properties and use of tinzaparin in oncology. Protamine sulfate partially (60% to 65%) neutralized tinzaparin’s anti-Xa activity. No dose adjustment of tinzaparin is needed even in patients with severe renal impairment and Creatinine Clearance ≥20 mL/min. Tinzaparin demonstrated a statistically significant decline in VTE recurrence at 1 year post the index thromboembolic event. A statistically significant reduction in minor bleeding rates was also described, whereas major bleeding events did not decrease in patients with cancer treated with tinzaparin versus those who received vitamin K antagonists. Tinzaparin treatment in patients suffering from deep vein thrombosis reduced the incidence of postthrombotic syndrome and venous ulcers. Tinzaparin’s ability to prevent both metastatic dissemination of cancer cells and tumor angiogenesis has been delineated in preclinical research. Current data show that tinzaparin is safe and efficacious either for short-term or for long-term treatment of VTE in patients with cancer. Clinical trials are needed in order to examine the utility of tinzaparin in primary prevention of VTE and validate its potential anticancer advantages exhibited in preclinical research.
Introduction
Cancer is not only a major health issue but also a growing economic burden for the whole world to deal with. Suggestive of its impact is the American Cancer Society’s estimate for a total of 1 688 780 new cancer cases and 600 920 cancer deaths in the United States in 2017. 1
In 1865, Trousseau was the first to observe what many studies have since proven: Patients with cancer, both hospitalized and those receiving outpatient chemotherapy, are at significantly increased risk of developing venous thromboembolism (VTE) compared to non-cancer patients. 2 –6 As a matter of fact, VTE occurs in up to 20% of patients with cancer. Likewise, the development of VTE in cancer betokens higher rates of recurrent VTE, bleeding complications due to anticoagulation therapy, morbidity, and mortality in comparison with the general population. 7 –11
A variety of risk factors are considered to be responsible for the higher prevalence of VTE in patients with cancer. These factors can be classified as patient, disease, and treatment related. Patient-related risk factors include advanced age, poor performance status, prothrombotic mutations, prior VTE, elevated platelet count before anticancer treatment, obesity, and comorbidities consisting of infections, renal disease, or heart failure. 12 –14 As far as disease-related factors are concerned, the presence of a tumor itself appears to cause a state of hypercoagulation involving procoagulant factors, tumor-derived cytokines, and direct interaction with a variety of cells. Primary cancer site is also deemed a risk factor for VTE development, with pancreatic and gastric cancers exhibiting the highest VTE rates, followed by primary malignant brain tumors, ovarian carcinomas, lung carcinomas, kidney carcinomas, and hematological malignancies. Patients with advanced stage and those with newly diagnosed malignancies are also at significantly higher risk of suffering from VTE. 14 –17 Accordingly, major surgery, prolonged immobilization, hospitalization, chemotherapy, adjuvant hormone therapy, antiangiogenic agents, erythropoiesis-stimulating agents, and the placement of central venous catheters comprise treatment-related risk factors. 14,17
Current guidelines recommend low-molecular-weight heparin (LMWH) treatment for the initial 5 to 10 days in patients with cancer with established VTE, as well as for secondary prevention of recurrence for at least 6 months. 18 –20 The use of LMWH or unfractionated heparin (UFH) is also recommended in patients undergoing major cancer surgery as a measure of primary prevention for up to 4 weeks after the procedure. 21 –26 Low-molecular-weight heparin, UFH, or fondaparinux should be administered for the prophylaxis of hospitalized patients with cancer with major medical illness or reduced mobility. 27 –29 In contrast, routine prophylaxis in ambulatory patients with cancer is not recommended, but prophylactic LMWH administration may be considered in ambulatory patients with high-risk cancer on a case-by-case basis. 30 In exception, patients with multiple myeloma receiving thalidomide or lenalidomide plus dexamethasone and/or chemotherapy are considered to have a VTE risk high enough to justify routine thromboprophylaxis with LMWH, warfarin (international normalized ratio [INR]: ∼1.5), or aspirin. 18 –20,31,32
As pivotal as their role in VTE therapeutics may be, not all LMWHs are the same. The purpose of this review is to study the pharmacological properties of tinzaparin, resulting in distinct clinical outcomes, and subsequently to examine the effectiveness of tinzaparin in the prophylaxis and treatment of cancer-related VTE, as well as its potential to alter the course of cancer disease.
Methods
A comprehensive search of PubMed and ScienceDirect databases was performed up to December 2016 using the keywords “tinzaparin AND (cancer OR oncology).” Specifically, eligible articles were those presenting original data from cross-sectional or longitudinal studies in adults or animals providing evidence on the pharmacological profile of tinzaparin, its use in patients with cancer, as well as its potential anticancer effects. The abstracts were screened to determine which studies and review articles were relevant to our objectives. Once duplicates were recognized and removed, the retrieved articles were then reviewed by 2 separate authors for inclusion or exclusion. Once all articles to be included were selected, the references of all included articles were reviewed to identify any additional applicable publications that may have been missed by the digital search. References from these articles were also obtained, and review articles are cited to provide readers with more details than this review has room for.
Results
The overall search identified 88 potentially relevant publications. Thirty-four articles reported data from original studies and were included in this review.
Overview of Pharmacological Profile
The enzymatic depolymerization of UFH from porcine intestinal mucosa via the utilization of Flavobacterium heparinum heparinase produces tinzaparin sodium (Innohep), a LMWH with an average molecular mass of 6500 Da (varying from 5500 to 7500 Da). 33 –35
Tinzaparin sodium demonstrates a dose-dependent, and greater compared to its anti-IIa activity, Xa inhibitory effect. 36,37 Tinzaparin also disposes the highest anti-IIa activity among all LMWHs. Thus, it has the lowest (2:1) anti-Xa/anti-IIa activity ratio, compared to bemiparin’s ratio of 8:1 and enoxaparin’s ratio of 3.9:1. 38,39
Data from in vitro as well as in vivo studies conclude that tinzaparin’s anti-Xa activity can be partially neutralized after protamine sulfate addition. Besides, among all LMWHs, tinzaparin demonstrates the highest rates of anti-Xa reversal in response to protamine sulfate. 40,41
Tinzaparin sodium results in a swift (in less than 1 hour) and sustained (lasting up to 5 hours) 2- to 5-fold elevation in plasma tissue factor pathway inhibitor (TFPI) levels. 42,43 Tinzaparin stimulates an increased release of free and total TFPI compared to bemiparin 44 ; conversely, no differences have been spotted in TFPI release versus enoxaparin. 45
It also produces a slight, but existent prolongation of the activated partial thromboplastin time (APTT) within normal range. 36,37 However, it appears to cause a significantly higher prolongation compared to bemiparin 39 and enoxaparin. 46 Neither hemoglobin levels nor platelet counts seem to be affected after tinzaparin administration. 37,47
Assuming its anti-Xa and anti-IIa activities as biomarkers, the pharmacokinetic parameters of tinzaparin sodium have been determined, using data from several studies. 36,48 Fossler et al conducted a randomized crossover study, involving 30 healthy volunteers. They measured an absolute bioavailability (F) of 86.7% (90% confidence interval [CI], 78.7%-95.5%) by comparing the mean anti-Xa AUC0→∞ of the subcutaneous formulation without preservative with that for intravenous (IV) administration of the same dosage (4500 anti-Xa IU). 36 The absolute F values of bemiparin (96%) 39 and enoxaparin (91%) 49 indicate a significant increase only in the latter case. The volume of distribution (V), computed again on the basis of anti-Xa activity after IV injection, ranged from 3.08 to 4.96 lt, connoting tinzaparin’s distribution to the vascular compartment, 36 in accordance with bemiparin and enoxaparin. 39,49
Finally, in contrast to other LMWHs, tinzaparin employs first-order pharmacokinetics with the consecutive involvement of cellular and renal route of elimination, exhibiting no bioaccumulation even in patients with severe renal impairment. Consequently, tinzaparin displays a clearance (Cl) of 1.14 to 2.66 lt/h, 36,41,48,50,51 whereas bemiparin and enoxaparin trail with 0.9 lt/h 52 and 0.64 to 1.33 lt/h, 53,54 respectively. It also demonstrates an elimination half-life (t1/2) of 3.41 to 4.13 hours after subcutaneous administration, which, compared to its t1/2 of 1.60 hours after IV administration, implies that the absorption of tinzaparin is slower than its elimination. Bemiparin has a t1/2 of 5.3 hours (the longest among all LMWHs), 39 while enoxaparin’s t1/2 is estimated at approximately 4 to 5 hours. 49
Evidence in Patients With Cancer
Venous thromboembolism treatment
The first study assessing the role of tinzaparin in patients with cancer was conducted by Hull et al in 2006. 55 In this multicenter, open-label, randomized study, 200 patients with cancer and proximal vein thrombosis were assigned to receive either tinzaparin in a therapeutic dose (175 IU/kg, subcutaneously [SC], once daily) for 12 weeks or UFH (5000 IU or 80 IU/kg bolus IV, followed by continuous IV infusion modified according to the APTT, terminated on day 6) superseded by warfarin (initiated on day 1 at 5 to 10 mg, dose adjusted in order to maintain an INR of 2 to 3, finally as a single therapy on day 6) for the same period of time. The primary efficacy end point of VTE recurrence did not demonstrate significant difference at 3 months between the 2 arms of the study; at 12 months, patients treated with tinzaparin displayed a statistically significant decline in VTE recurrence (7% versus 16%; P = .044; risk ratio: 0.44; absolute difference −9.0%; 95% CI, −21.7% to −0.7%). All bleeding events during therapy, representing the primary safety end point, appeared in 27% of patients (7% major bleeding) treated with tinzaparin and 24% of patients (7% major bleeding) of those receiving UFH in combination with warfarin (P > .05; absolute difference −3.0%; 95% CI −9.1% to 15.1%). Neither 3-month nor 12-month mortality showed any survival benefit between the 2 groups. The incidence of thrombocytopenia at 3 months or bone fractures at 12 months did not vary significantly in the 2 groups of patients.
In 2012, Laporte et al 56 published a meta-analysis of 5 randomized controlled trials, intending to investigate matters of efficacy and safety of long-term curative doses of tinzaparin compared to vitamin K antagonists (VKAs; warfarin or acenocoumarol) both in the general population (n = 1662) and in patients with cancer only (n = 283). Patients with cancer exhibited a statistically nonsignificant (38%) relative risk (RR) reduction (RR = 0.62; 95% CI, 0.34-1.13; P = .12) at the end of the 3- to 6-month treatment period, which raised to 59%, becoming statistically significant at 1 year (RR = 0.41; 95% CI, 0.21-0.79; P = .008). Major bleeding (overt and associated with a decrease in hemoglobin of 2 g/dL or more; leading to a transfusion of 2 or more units of blood; retroperitoneal; intracranial; occurring in a major joint) rates as well as all-cause mortality at 3 to 6 months and at 1 year did not present significant differences among patients with cancer.
Lee et al 57 performed a multicenter, open-label, randomized clinical trial enrolling 900 adult patients with active cancer (histological diagnosis of cancer and receiving anticancer therapy or diagnosed with or received anticancer therapy within the previous 6 months), with objectively documented deep vein thrombosis (DVT) or pulmonary embolism, with a life expectancy of greater than 6 months, and without contraindications for anticoagulation. The patients were treated either with tinzaparin (175 IU/kg, SC, once daily) for 6 months or with tinzaparin (175 IU/kg, SC, once daily) for the first 5 to 10 days of treatment period, followed by warfarin (in an adjusted dose, so as to maintain the INR between 2 and 3) for an overall of 6 months. The study’s duration was 180 days, with follow-up of 30 days after the last medical dose. The VTE recurrence at 6 months occurred in 7.2% of patients (31 of 449) receiving tinzaparin versus 10.5% (45 of 451) of those receiving warfarin (hazard ratio [HR] = 0.65; 95% CI, 0.41-1.03; P = .07). No significant variation was marked between the 2 arms of the study, in regard to major bleeding rates, occurring in 12 patients treated with tinzaparin versus 11 patients treated with warfarin (HR = 0.89; 95% CI, 0.40-1.99; P = .77). However, a statistically significant decrease in clinically overt nonmajor bleeding rates was indicated; these complications appeared in 49 of 449 patients in the tinzaparin arm, compared to 69 of 451 patients in the warfarin arm (HR = 0.58; 95% CI, 0.40-0.84; P = .04). Finally, no significant difference in all-cause mortality was observed (150 patients for tinzaparin versus 138 for warfarin; HR = 1.08; 95% CI, 0.85-1.36; P = .54).
Venous thromboembolism prophylaxis
In a recent single-arm, open-label, pilot trial, Perry et al 52,58 evaluated the safety of prophylactic doses of tinzaparin (4500 IU administered SC, once daily, initiated between 48 hours to 4 weeks after the most recent surgical procedure) for a planned duration of 12 months. Forty patients with newly diagnosed, grade III to IV malignant glioma were enrolled. About 2 (5%) patients developed central nervous system hemorrhage, 1 grade I and 1 grade II. Only 1 patient suffered from DVT while receiving tinzaparin. Therefore, tinzaparin is considered to be safe for VTE prophylaxis in patients with brain tumor.
Anticancer Properties
Apart from its anticoagulant abilities, tinzaparin sodium possesses multiple de novo anticancer effects, as demonstrated in various preclinical models. Table 1 summarizes tinzaparin’s anticancer properties.
Anticancer Properties of Tinzaparin Sodium.
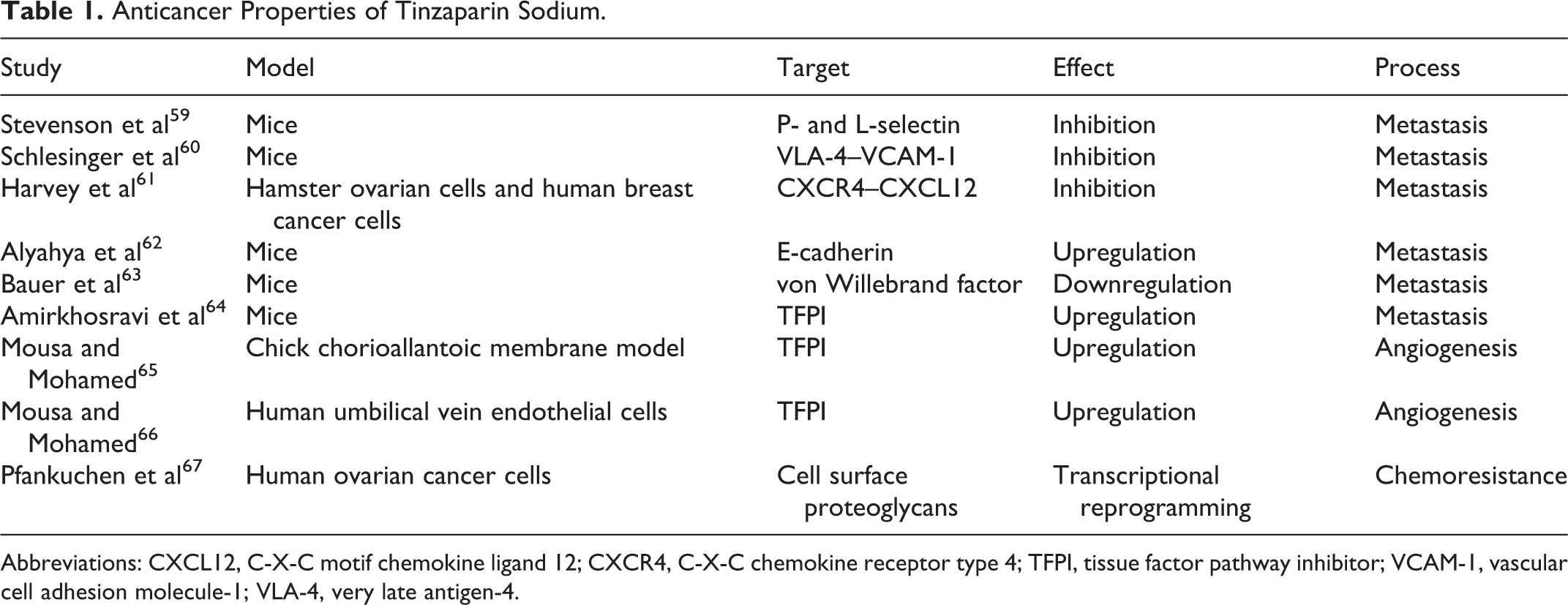
Abbreviations: CXCL12, C-X-C motif chemokine ligand 12; CXCR4, C-X-C chemokine receptor type 4; TFPI, tissue factor pathway inhibitor; VCAM-1, vascular cell adhesion molecule-1; VLA-4, very late antigen-4.
Local Tumor Growth
The activation of Extracellular signal-Regulated Kinase (ERK) pathway is an established mechanism that prompts cell division, enhancing tumor cell proliferation. 68,69 As a result, several anticancer drugs have been developed targeting the inhibition of this specific pathway. 68,70 –72 Tinzaparin, along with other LMWHs, has been shown to limit downstream phosphorylation of ERK kinase pathway. 73 However, tinzaparin failed to impede cellular proliferation in a model of in vitro human breast cancer cells. 74 Likewise, tinzaparin did not demonstrate any effect on primary tumor growth in an experimental B16F10 metastasis model. 75
Metastasis
As reviewed elsewhere, the most detrimental aspect of cancer disease, metastatic spreading, occurs in a series of subsequent steps, also known as the metastatic cascade. First of all, epithelial–mesenchymal transition ensues, causing loss of cell polarity, downregulation of E-cadherin paired with upregulation of N-cadherin expression, as well as acquisition of spindle cell morphology. This local and ephemeral cell transformation is capable but not always essential for the promotion of tumor cell migration (by the development of cell membrane bulges) and invasion (by derangement of the extracellular matrix [ECM]) surpassing the basal membrane barrier. Tumor cells should then overcome apoptosis induced due to inadequate cell adhesion to its surrounding cells or the ECM called anoikis. Anoikis regularly represents a homeostatic tissue mechanism. Angiogenesis is another critical step further aiding metastatic spread of malignant cells. In order to metastasize, tumor cells should then enter the circulation (intravasation), avoid once again their destruction by anoikis, the immune cells, or sheer stress from the blood flow, and manage to exit the circulation (extravasation) in a distant site. Disseminated cancer cells then form micrometastases. These last events require not only a favorable microenvironment at the target site but also the prevention of tumor cell dormancy or anoikis for 1 last time. 76 Regression to their former epithelial state by mesenchymal–epithelial transition is also fundamental for disseminated cancer cells to result in micrometastases and finally grow into macrometastases. 77
Platelet-assisted tumor cell adhesion to the vascular endothelial cell lining is a crucial incident in the intravasation as well as the extravasation process. 78 A variety of cellular interactions have been involved in this event. Stevenson et al concluded that clinically relevant doses of both UFH and tinzaparin diminished metastatic rates, via P- and L- selectin inhibition, in a mice metastasis model. 59 Furthermore, the antimetastatic effect of heparin is abolished in double P- and L-selectin-deficient mice. 79 In addition, Schlesinger et al 60,80 used very late antigen-4 knockdown (VLA-4kd) B16F10 murine melanoma cells to assess the role of VLA-4/vascular cell adhesion molecule-1 (VCAM-1) interaction in the metastatic process. The VLA-4kd cells’ loss of the ability to interact with VCAM-1 resulted in a reduced metastatic rate in mice, compared with control. However, VLA-4/VCAM-1 bridging blockade has a nonsignificant cumulative contribution to the establishment of metastatic foci in a P-selectin-deficient background, both in vitro and in vivo, indicating that selectin-mediated interactions prevail over integrin-mediated ones. Tinzaparin administration in both B16F10- and B16F10-VLA-4kd-injected mice significantly reduced metastatic rates. Thus, tinzaparin displays a role in disturbing not only P- and L-selectin but also VLA-4/VCAM-1 interconnections in vivo. Tinzaparin also inhibited C-X-C chemokine receptor type 4-expressing malignant cells binding to C-X-C motif chemokine ligand 12 on normal tissue, resulting in a significant decline in metastatic dissemination of human breast cancer cells to the lung in a murine model. 61
A pancreatic cancer mouse model has demonstrated tinzaparin’s role in upregulating the expression of E-cadherin in malignant cells. Besides, depressed E-cadherin expression increases local invasion and migration, further promoting metastasis. 62
Extracellular matrix degradation represents another step toward metastatic dispersion of malignant cells. Heparanase is a proteolytic enzyme-mediating ECM degeneration. 81,82 Although there are no studies hitherto examining the potential of tinzaparin in suppressing heparanase, tinzaparin represents the most powerful aggrecanase inhibitor. 83 Aggrecanase-1 or ADAMTS4 and aggrecanase-2 or ADAMTS5 are proteolytic enzymes also accounted for ECM disruption. A number of studies link both aggrecanases to the formation of several types of solid tumors. 84,85
Tumor cells activate nearby endothelial cells by tumor-derived vascular endothelial growth factor-A (VEGF-A). Endothelial cell stimulation causes von Willebrand factor (vWF) release, which coupled with local suppression of both the expression and the proteolytic activity of ADAMTS13 establishes a procoagulatory microenvironment inside the tumor vasculature. Additionally, vWF clusters attract and bind platelets, which in turn protect tumor cells from immune cells or directly promote their extravasation, an essential step in the metastatic cascade. Bauer et al demonstrated that tinzaparin injection impeded tumor progression and improved survival in Ret transgenic mice. 63
The antimetastatic effect of tinzaparin has also been projected in a B16 melanoma cell lung metastasis model in mice. Subcutaneous administration of tinzaparin 4 hours prior to the IV infusion of melanoma cells induced lung tumor formation by 89% compared with controls (P < .001). Additional daily administration of tinzaparin for 14 days after the initial dose achieved a further reduction in lung tumor formation, reaching a rate of 96%. 64
Angiogenesis
Angiogenesis, as mentioned before, is a cardinal step in local tumor growth and metastatic progression. Solid tumors generate themselves a propitious proangiogenic background. Tumor-induced angiogenesis can be triggered in a hypoxia-induced factor-1 (HIF-1)-dependent or -independent manner. Rapid malignant cell proliferation to form a solid tumor impairs the local balance between oxygen supply and demand, causing intratumoral hypoxia, which in turn stimulates HIF-1 production. In addition, carcinogenic genetic alterations in oncogenes as well as in tumor suppressor genes provide the essential stimulus for the increased output of HIF-1 or its decreased proteasomal degradation. Thus, HIF-1 accumulation upregulates VEGF-a expression, resulting in angiogenesis. Other mechanisms, such as the Warburg effect (a shift to anaerobic glucose metabolism, causing an increase in lactate and pyruvate concentrations), implicated in the induction of HIF-1-dependent angiogenesis. 86 On the other hand, HIF-1-independent mechanisms of angiogenesis in patients with cancer involve multiple RAS signal transduction pathways. These pathways stimulate not only VEGF production but also the release of other proangiogenic mediators such as interleukin-8, CXCL1, and Prostaglandin E2. 87 Apart from VEGF-a, other factors known to trigger the “angiogenic switch” include fibroblast growth factors, platelet-derived growth factor, and epidermal growth factor. Conversely, thrombospondin 1, angiostatin, endostatin, and tumstatin rather impede the angiogenic process.
Malignant cells can also incite the spontaneous or hypoxia-driven synthesis and assembly of tissue factor (TF) factor VII complexes. Cleavage of Protease Activated Receptor 2 by the above complex promotes downstream VEGF-a and other proangiogenic factors production, further assisting in tumor-derived angiogenesis. 88 Tinzaparin possesses an antiangiogenic potential mediated via TFPI release. Mousa and Mohamed 65 indicated that this potential is dose dependent, but stimulus independent both in vitro and in vivo in a chick chorioallantoic membrane tumor implant model. Tinzaparin and recombinant TFPI reduced the growth of colon carcinoma, human fibrosarcoma, and human lung carcinoma tumors in the above model. This ability could be reversed by a specific anti-TFPI monoclonal antibody. A human umbilical vein endothelial cell angiogenesis 66 model came to confirm the previously stated antiangiogenic ability of tinzaparin. Furthermore, this model added that this ability is correlated with TFPI release but not with tinzaparin’s anti-Xa activity.
Reversal of Chemoresistance
In 2015, Pfankuchen et al 67 revealed a pioneering aspect of tinzaparin’s anticancer properties; using tinzaparin in a dose corresponding to its therapeutic antithrombotic dosage in adults, they managed to reverse cisplatin resistance of A2780 human ovarian cancer cell lines. Experimental data ruled out increased intracellular uptake of tinzaparin among chemoresistant cells. Nevertheless, tinzaparin’s interaction with heparan sulfate proteoglycans (HSPGs) on the cell surface and downstream HSPG signaling resulted in alteration of the expression of 3776 genes in A2780 cisplatin-resistant cells. Hence, tinzaparin seems to bear an impact on various cell systems, explaining its newly found properties.
Discussion
The use of LMWH is currently considered as mainstay for the treatment and secondary prevention of cancer-associated VTE. Among all LMWHs, tinzaparin possesses the lowest anti-IIa/anti-Xa activity ratio (2:1). The first step for the LMWHs in order to exert their action is to form a complex with antithrombin (ATIII), binding it to their unique pentasaccharide sequence. After bonding, a structural change in ATIII occurs, resulting in a 1000-fold increase in its ability to interact with factor Xa. 89 –91 In contrast, ATIII-mediated factor IIa (thrombin) inhibition requires the formation of a ternary heparin–antithrombin–thrombin cluster. This cluster can be assembled only in 18-saccharide long LMWH chains, considerably limiting LMWHs’ anti-IIa potential, since only 25% to 50% of their chains meet this prerequisite. 89,92,93
Tinzaparin sodium’s Xa inhibitory effect can be partly neutralized by the use of protamine sulfate. Data from an in vitro study conclude that 85.7% of tinzaparin’s anti-Xa activity is neutralized after protamine sulfate addition, compared to a lesser extent of neutralization among the other LMWHs. 40 These data are in correspondence with another study in 50 healthy volunteers, a nonrandomized this time, in which Holst et al came to the conclusion that protamine sulfate reversed 80% and 60% to 65% of tinzaparin’s anti-Xa activity following IV or SC injection, respectively. The 65% to 75% return of anti-Xa activity seen in the SC groups 3 hours after the reversal marks the continuous absorption of the LMWH from the SC depot, rather than the insufficient dosage of the antidote. 41 The main reason for the varying degree of anti-Xa reversal exhibited by different LMWHs seems to be a combination of both the molecular size of the LMWH chains and their sulfate charge density.
In addition, tinzaparin sodium administration promotes TFPI release. The TFPI inhibits the factor VII–TF complex, modulating the initiation of coagulation induced by TF. It also directly inhibits factor Xa. 94,95 As a result, part of the LMWH anticoagulant potential is believed to be mediated via endothelial TFPI release. 96 –99 Besides, low total and free TFPI plasma levels constitute a risk factor for DVT. 100
Both animal 101 and human 102 studies have demonstrated the dose-dependent pharmacokinetics of UFH. Its elimination is best described as a combination of 2 systems. Cellular uptake (reticuloendothelial system, endothelial cells), mediated via hyaluronic acid receptor for endocytosis receptors, 103 is saturable and more efficient at low-dose range, whereas renal excretion, representing an active tubular process, is nonsaturable and becomes prevalent as doses increase. The above concept is typically less conspicuous in LMWHs, due to its molecular weight (MW) dependency. 104 –107 Hence, LMWHs with a MW below approximately 5000 Da (such as bemiparin, enoxaparin, nadroparin, and so on) are predominantly excreted by the kidney, in a dose-independent manner. On the contrary, tinzaparin (6500 Da) and to a lesser extent dalteparin (5700 Da) employ first-order pharmacokinetics, with the involvement of cellular and renal routes of elimination successively. 108 Accordingly, in patients with mild-to-severe renal impairment, defined as Creatinine Clearance - CrCl ≥20 mL/min, prophylactic dosage (4500 anti-Xa IU) of tinzaparin does not accumulate; thus, tinzaparin administration requires no dose adjustment in this setting. On the other hand, bemiparin, enoxaparin, and certoparin do accumulate demanding dose reduction; no data were available for nadroparin. 109,110 In the same subgroup of patients, as far as therapeutic dosage (175 anti-Xa IU/kg) is concerned, tinzaparin continues to show no accumulation, so no dose adjustment is currently recommended. 111 –119 Enoxaparin continues to display bioaccumulation, requiring dose reduction. 113 Sufficient data are lacking in the case of dalteparin. 111,120 This conclusion appears paramount for the subpopulation of patients with cancer, as they combine multiple factors that aggravate their renal function, such as older age, dehydration, and use of nephrotoxic agents for anticancer treatment, and other comorbidities, such as hypertension, diabetes mellitus, and so on.
Hull et al 55 were the first to report the statistically significant benefit in VTE recurrence at 12 months post the index thromboembolic event for patients treated with tinzaparin for 12 weeks, in comparison with those who received VKAs for the same period of time. In this study, outcomes were evaluated both at the end of treatment period and 12 months, since there is clinical evidence implicating that heparin and its low MW fragments maintain their beneficial effect even after cessation of therapy. 121,122 Neither bleeding events nor mortality exhibited statistically significant difference between the 2 arms of the study.
Laporte et al’s meta-analysis 56 came to confirm the aforementioned outcomes concerning patients with cancer. In contrast, no difference was marked when tinzaparin was administered for the treatment of VTE in the general population neither at the end of treatment period nor at 1 year, as compared to VKAs.
The above results come in accordance with the CLOT study, 123 which investigated the role of dalteparin in cancer-related VTE. Conflicting data 124 –127 exist in the case of enoxaparin as VTE anticoagulation treatment.
As far as matters of safety are concerned, the CATCH trial outlined tinzaparin’s better tolerated profile, in terms of a statistically significant reduction in overt nonmajor bleeding rates, in comparison with VKAs for the treatment of acute VTE in patients with cancer; tinzaparin use did not result in decreased major bleeding events. 57
However, Noel-Savina et al 128 failed to confirm any correlation between the choice of anticoagulant and the risk of recurrent VTE. Instead, this retrospective cohort study involving 250 patients with cancer concluded that early (before 6 months) cessation of anticoagulation therapy either in patients at low risk 58 of recurrence or for a reason other than bleeding or death represented the only factor related to a statistically significant elevated risk of VTE recurrence (OR = 7.2; 95% CI, 2.0 to 25.7; P = .002). Indeed, the risk was 8-fold higher when anticoagulation stopped before 6 months.
Although many clinical trials have highlighted the role of tinzaparin in the acute treatment as well as the secondary prevention of cancer-related VTE, its role in primary prevention has not been as much documented. 52
Postthrombotic syndrome (PTS) represents a frequent, wearing, and costly long-term complication of VTE. Daskalopoulos et al 129 were the first to report tinzaparin’s superiority over VKAs in reducing the incidence of PTS and venous ulcers. The Home-LITE trial confirmed a statistically significant decline of the previously stated events in the tinzaparin group, compared to those treated with oral VKAs. 130 In addition, tinzaparin exhibited greater rates of recanalization of leg thrombi as compared to VKAs. Although the above results were registered in the general population, the pathophysiology of PTS remains unchanged in cancer-induced VTE. Prolonged overall survival due to advances in cancer therapeutics also increases the prevalence of PTS in patients with cancer.
Furthermore, sufficient preclinical data, including known pathophysiologic mechanisms and both in vitro and in vivo animal studies, have revealed tinzaparin’s anticancer properties. As reviewed elsewhere, tinzaparin has displayed both antimetastatic and anti-angiogenic abilities. Another question that remains to be answered is whether preclinical evidence can be translated in clinical outcomes, in terms of increased overall survival by adding tinzaparin to standard chemotherapy regimens?
In this setting, Auer et al 131 conducted a randomized, controlled, pilot study, involving 18 patients with localized and resectable colon cancer. These patients received standard (4500 IU, SC, once daily, initiated 8 hours after surgery and terminated on the day of discharge) or extended (4500 IU, SC, once daily, initiated 8 hours and terminated 4 weeks after surgery) perioperative thromboprophylaxis. The primary goal was recruitment rate. The secondary goals consisted of compliance with therapy, major and minor bleeding rates, and finally disease recurrence. Excellent compliance with tinzaparin injections, a total of 2 (11%) major bleeding events and only 2 (11%) patients with recurrent disease, concluded that a large, multicenter, randomized clinical trial exploring disease-free survival in patients with resectable colon cancer is both safe and feasible.
Overall survival is the primary end point of the TILT study, 132 a randomized controlled clinical trial, enrolling patients with completely resected stage I, II, or III (T3N1) lung cancer.Patients are divided into 2 groups: control group and experimental; those in control group will receive usual postoperative care, while patients in the experimental 1 will receive the usual postoperative care, plus tinzaparin (100 IU/kg, SC, once daily, for 90 days). Follow-up period will last for 3 to 8 years. Finally, the avant-garde ability of tinzaparin to reverse chemoresistance of ovarian cancer cells to cisplatin in vitro remains to be confirmed in vivo. Another interesting prospect is whether tinzaparin can reproduce this ability in human cancer cells with acquired resistance to anticancer agents other than cisplatin.
Conclusion
Tinzaparin sodium possesses important pharmacodynamic and pharmacokinetic properties, maintaining an exceptional stand among other LMWHs. The LMWH prescription constitutes standard of care for the treatment and secondary prevention of VTE in oncology. Tinzaparin administration has demonstrated substantial benefits over VKAs in matters of efficacy supplementary to safety for the treatment of venous thromboembolic events in patients with cancer. Its innate anticancer effects have also been delineated in preclinical research. Head-to-head studies are needed in order to investigate whether tinzaparin’s unique pharmacological properties can be translated in lower rates of VTE recurrence, along with fewer bleeding events in patients with cancer. Randomized controlled clinical trials are eventually required to investigate its role in primary cancer-associated VTE prevention and validate its ability to alter the course of cancer disease.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.