
Abstract
Hemophilia A (HA) is an X-linked bleeding disorder caused by heterogeneous mutations in the factor VIII gene (F8). Our aim is to identify the causative mutations in a large HA cohort from China. We studied 216 unrelated HA families. Molecular analyses of F8 were performed using a combination of molecular techniques, including polymerase chain reaction, direct sequencing, and multiplex ligation-dependent probe amplification. The deleterious consequences of the unreported missense mutations were evaluated using various bioinformatics approaches. Causative mutations in F8 were identified in 209 families, intron 22 inversion (Inv22) was identified in 89 severe families, and intron 1 inversion (Inv1) was positive in 5 severe families; 95 mutations were detected among 115 noninversion families, of which 42 were novel, including 29 null variations and 13 missense mutations for which causality was demonstrated via bioinformatics. Among the 53 previously reported mutations, more nonsense (5 of 9) and missense (10 of 26) mutation sites were found to occur at Arginine (Arg) sites and multiple small deletions/insertions (5 of 10) located within the poly-A runs of the B domain. The majority of these sequence variants frequently recurred in the database. The odds ratios for the likelihood of developing inhibitors significantly increased in the presence of nonsense mutation. Our F8 defect spectrum was heterogeneous. Small deletions/insertions in the poly-A runs of the B domain and nonsense and missense mutations at Arg sites were identified as mutation hot spots. Nonsense mutation increased the risk of developing inhibitors.
Introduction
Hemophilia A (HA; OMIM 306700) is an X-linked hereditary disease caused by a deficiency in coagulation factor VIII (FVIII) and occurs at a frequency of 1 of 5000 live-born males. 1 According to the baseline FVIII coagulant activity (FVIII: C) level present in the blood of an affected individual, HA may be classified as severe (<1%), moderate (1%-5%), and mild (5%-40%). 2 The development of inhibitors in patients with HA presents a major complication for treatments involving FVIII. 3 The FVIII gene (F8) is located on the distal end of the long arm of the X-chromosome (Xq28); this gene spans 186 kb of genomic DNA and is divided into 26 exons (approximately 9 kb of complementary DNA [cDNA]). F8 is translated into a 2351-amino acid polypeptide chain with a 19-amino acid leader peptide. The mature peptide comprises structural domains that are referred to as A1-A2-B-A3-C1-C2 and is activated following the dissociation of the B domain. 4 Circulating FVIII in the blood is bound to the von Willebrand factor (vWF), which protects FVIII against degradation. Typically, the most common gene defects associated with severe HA are intron 22 inversion (Inv22) and intron 1 inversion (Inv1), which occur in 40% to 50% 5 and 2% to 5% 6 of patients with HA with the severe disease phenotype, respectively. In the remaining patients with HA, a large and heterogeneous spectrum of F8 gene mutations has been reported in the Hemophilia A Mutation, Structure, Test and Resource Site (HAMSTeRS; http://hadb.org.uk), including a variety of point mutations that results in amino acid replacements (missense), premature termination codons (nonsense), or RNA splice-site defects, as well as deletions, duplications, and insertions.
In our study, we recruited 216 unrelated Chinese HA families that constitute approximately one-half of the registered patients with HA at the Shanxi hemophilia center. Many of the patients receive only on-demand treatment and have disabling hemophilic arthropathy. Hemophilia is still a crippling and life-threatening disease in China. Therefore, we have encountered a need for the family to receive genetic counseling. For carrier detection, we need index patients. Our study focuses on the genetic diagnoses and F8 mutation spectrum analyses in this large HA cohort.
Methods
Participants
We studied 223 Chinese patients with HA from 216 apparently unrelated families (186 severe, 14 moderate, and 16 mild cases) diagnosed between 2010 and 2014. All but one of the patients were male, and their age at the time of recruitment ranged from 1 to 63 years. The FVIII: C levels were measured by standard 1-stage clotting assay using commercially available FVIII-deficient plasma (Dade Behring, Marburg, Germany). The FVIII inhibitor titers were quantified using the Nijmegen modification of the Bethesda assay. 7 A total of 100 randomly selected healthy male controls were recruited to test whether the novel missense mutations present in the HA cohort were neutral single-nucleotide polymorphisms (SNPs). All of the patients or their guardians provided informed consent for the molecular studies. Ethical approval for the study was obtained from the Medical Ethical Committee of the Second Hospital of Shanxi Medical University.
Molecular Genetic Analyses
Genomic DNA was extracted from 200 µL of peripheral blood using the QIAamp DNA Mini Kit (Qiagen Inc, Valencia, California, USA), according to the manufacturer’s instructions. First, we screened for the presence of Inv22 using the kit provided by Yaneng Bioscience Co, Ltd (Shenzhen, China) via long-distance polymerase chain reaction (PCR). 8 The negative patients were then tested for Inv1 via multiplex PCR. 6 DNAs from those patients with severe HA lacking either inversion were subjected to a PCR amplification analysis of the essential F8 regions (promoter, exons, splice junctions, and 3′ polyadenylation signal region) according to a previously reported protocol in HAMSTeRS (http://hadb.org.uk/WebPages/Database/Methods/pcr.html), and the amplified DNA fragments were then sequenced by BGI Tech (Beijing, China). We explored the possibility that the novel missense mutations might have been neutral SNPs by screening for each in a cohort of 100 normal male patients. When the PCR amplification products were consistently absent in 1 or more exons or no identified mutations were detected, the probable large deletions and duplications were examined using a multiplex ligation-dependent probe amplification (MLPA) assay 9 with the SALSA MLPA probemix P178-B2 F8 (MRC-Holland, Amsterdam, the Netherlands) at Sangon Biotech (Shanghai, China).
At the cDNA level, the nomenclature of the detected sequence variations was described according to HAMSTeRS, with the “A” of the ATG codon for translation initiation numbered + 1. At the protein level, the first methionine was numbered as −19, based on the mature protein.
Prediction Software
Amino acid conservation in the human, porcine, murine, and canine species was represented according to HAMSTeRS (http://hadb.org.uk/WebPages/Database/Protein/lineups.html). Two commonly used algorithms to predict the possible impact of an amino acid substitution on the protein structure and function were employed: polymorphism phenotyping (PolyPhen-2, http://genetics.bwh.harvard.edu/pph2/,SupplementarySoftware) and sorting intolerant from tolerant (SIFT, http://sift.jcvi.org/www/SIFT_aligned_seqs_submit.html). The 3-dimensional structure of FVIII was visualized using the PyMOL Molecular Graphics System (http://www.pymol.org). The GRASP2 program was used for further analysis of each macromolecular domain structure of the protein and for electrostatic surface visualization. 10 The secondary structure of the in-frame deletion was studied via the PSIPRED algorithm (http://bioinf.cs.ucl.ac.uk/psipred/). 11
Statistical Analysis
The odds ratios (ORs) with 95% confidence intervals for inhibitor development in each F8 mutation category were calculated using SAS software, version 9.2 (SAS Institute Inc, Cary, North Carolina, USA). Comparisons were made using Fisher exact test and were considered significant if the P value was less than .05.
Results and Discussion
Mutation Spectrum
Following the molecular diagnostic workflow, disease-causing F8 mutations were identified in 209 of the 216 unrelated families, representing a mutation detection rate of 96.8%. The distribution of different categories of F8 mutations identified is shown in Table 1. Aside from Inv22, the most frequent mutation type resulting in severe hemophilia was point mutation (24.7%, 46 of 186), including missense mutation, nonsense mutation, and splice-site changes; this was followed by small deletion/insertion mutations (15.6%, 29 of 186). The prevalence was compatible with the rates observed in previous studies, 12 –14 and as expected, all of the null mutations were associated with the severe hemophilia, whereas missense mutations were responsible for all levels of HA severity. Despite using multiple techniques, no mutations were identified in the remaining 7 severe individuals; in these cases, the mutations may have been located deep within introns or in regions outside of F8 that were important for its expression. 15 Nevertheless, it is also possible that these patients had type 2N von Willebrand disease (tyepe 2N vWD).
Type of F8 Gene Mutation in 216 Chinese Families With HA.

Abbreviations: F8, factor VIII gene; HA, hemophilia A.
Large Rearrangements
The prevalence rates of Inv22 and Inv1 in our population were consistent with those reported worldwide. 16 –19 In 1 sporadic family with only 1 affected Inv22 individual, the mother was found to be a carrier; however, this inversion was not present in either maternal grandparent, which coincides with the hypothesis that the inversion may have occurred during male meiosis. 16 Despite this finding, a noncarrier mother has an affected son with Inv1 but a negative family history, a maternal origin for the inversion is indicated. One obligate carrier with the severe HA phenotype was heterozygous for Inv22. Given the absence of a second mutation after screening the entire gene, an extremely skewed inactivation of the X-chromosome bearing a normal F8 allele is the most likely mechanism for this presentation. 20
Ten different large rearrangements, including 9 large deletions and 1 large duplication spanning exons 13 to 25, were detected via the MLPA analysis. In 5 families, only a single-exon deletion was identified. These deletions involved exons 1, 3, 14, 15, and 24. The absence of a signal for multiple exon 1, 3, and 14 probes excluded the presence of a nucleotide mismatch at the probe-binding site. Other deletions affecting only single exons, including exons 15 and 24, were not confirmed via the recommended quantitative PCR-based messenger RNA evaluation. 21 Four other index cases exhibited larger deletions of exons 2 to 6, 7 to 8, 6 to 12, and 25 to 26, the last of which extended to the 3′-untranslated region (UTR). The deletions involving exons 1, 15, and 2 to 6 have already been reported in HAMSTeRS and in the recent literature. 22 The patient with a large deletion encompassing exons 2 to 6 developed an inhibitor. The inth1h homolog, which exists in the opposite orientation, may allow a nonallelic homologous recombination mechanism that results in the deletion of exon 1. 23
Small Deletions/Insertions
All 24 of the small deletion/insertion mutations (16 deletions and 8 insertions) that were identified in 29 families with the severe phenotype are summarized in Table 2. Overall, 79.2% (19 of 24) of these mutations (13 of 16 deletions and 6 of 8 insertions) occurred in exon 14. Among the 10 previously reported small deletions/insertions, 4 insertions and 1 deletion from 8 unrelated families were identified within long poly-A runs (defined as at least 6 consecutive adenines) in exon 14, which is a hot spot for such mutations. 24 Within a sequence of 9 adenines in exon 14, a deletion (c.3637delA) was identified in 3 unrelated severe HA families and an insertion (c.3637insA) was detected in 2 unrelated severe HA families; thus, the single-nucleotide deletion/insertion in this site demonstrated the presence of hot spot mutations in our cohort. All but 1 of our patients with insertion and deletion mutations resulted in frameshifts leading to premature stop codons. The exception is one with a 3-base deletion (c.2014-2016delTTC mutation) that caused an in-frame amino acid deletion (p.Phe653del). The secondary structure of this locus was changed to a combination of coiled and β-strands instead of only β-strands as in wild-type FVIII (identified using PSIPRED). This change caused a loose spatial protein conformation, disrupted the packing buried hydrophobic side chains, and thereby affected the folding or stability of the A2 domain subunit. 25
Description of Insertions/Duplications and Deletions Detected in the Patients Included in This Study.a
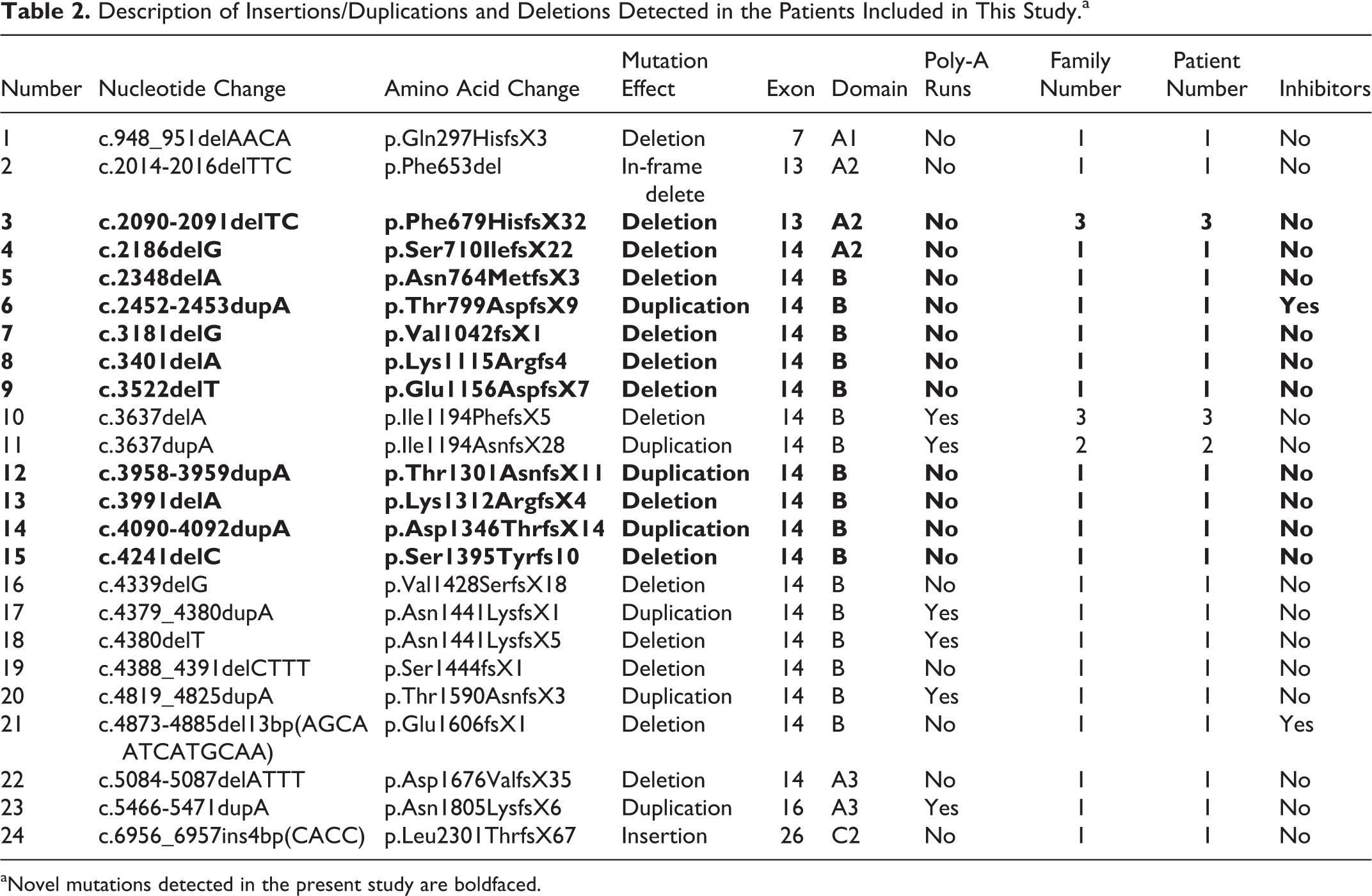
aNovel mutations detected in the present study are boldfaced.
Single-Nucleotide Substitutions
A total of 61 point mutations were identified in our study, including 39 missense, 15 nonsense, and 7 splice-site mutations; a detailed description of these mutations is provided in Table 3. Thirty-nine missense mutations were identified in our 52 HA families (22 severe, 14 moderate, and 16 mild). In agreement with other reports, 25,26 the majority of the previously reported nonsense (5 of 9) and missense (10 of 26) mutations in our cohort occurred in Arg. All of these identified genetic variants had been reported in HAMSTeRS frequently (the 10 above-reported cases), indicating that Arg is a hot spot for nonsense and missense mutations.
Detailed Description of Point Mutations Detected in Our Patients.a
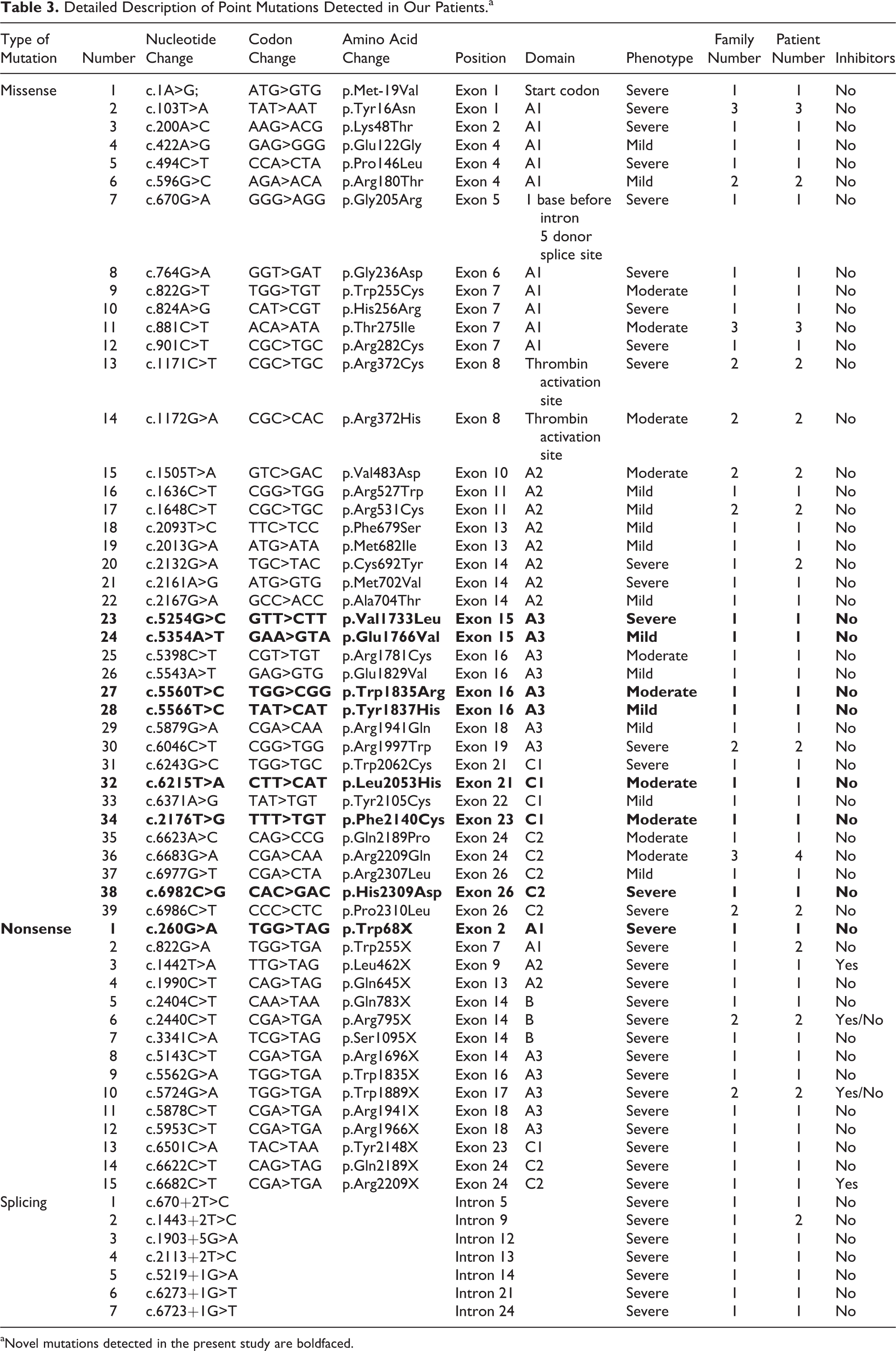
aNovel mutations detected in the present study are boldfaced.
Our series detected 6 single-nucleotide substitutions in the B domain, a region that lacks procoagulant activity and is partially spliced from the mature protein. Four of the mutations had been reported previously, p.Asp1241Glu and p.Ser1269Ser were reported as SNPs, and both p.Pro928Arg and p.Val1492Ile were registered in HAMSTeRS as disease-causing mutations of patients with severe HA who also carried Inv22. The transient expression result of p.Pro928Arg variation in HEK293T cells indicates that this mutation might cause a mild HA phenotype. 27 However, a male in our study with p.Pro928Arg mutation has normal FVIII activity, suggesting that this represents a rare SNP. The p.Val1492Ile mutation was identified in our severe patient with a large deletion involving exon 15; a novel amino acid–changing variation of p.Met779Thr was detected in our severe patient along with a large deletion including exon 3, whereas a novel silent c.4527A>C (p.Gly1490Gly) mutation was observed in a severe patient with Inv22. The presence of additional causative mutations might explain the severe phenotypes observed in these 3 patients, indicating the 3 genetic variations in the B domain are rare polymorphisms rather than causative mutations. In addition to the SNPs described above, the frequencies of the variant A allele of the SNPs in intron 7 (c.1010-27G>A) and in the 3′-UTR (c.8270G>A) were 5.6% and 1.6%, respectively.
Novel Mutations
Of the 95 different mutations detected in our study, 42 genetic alterations in F8 were neither identified in the HAMSTeRS nor reported in recent publications. These novel mutations occurred at a significant frequency (44.2%), indicating that our patients exhibit a specific mutational profile when compared with the previously reported data. A total of 29 novel molecular defects were predictive of null variants, including 6 nonsense mutations, 14 frameshift mutations, 6 large deletions, 1 large duplication, and 2 splice-site changes (c.670+2T>C and c.5219+1G>A) that destroyed the conserved GT of the donor splice junction. The remaining 13 novel missense mutations were not found in the 100 normal male individuals, the available population SNP databases, the National Center for Biotechnology Information (http://www.ncbi.nlm.nih.gov/projects/SNP/), or the International HapMap Project (http://www.hapmap.org/), suggesting nonpathogenic polymorphism is not likely. When compared to the HAMSTeRS database, 9 novel missense mutations were located at residues that had been previously related to HA; however, our mutations harbored different amino acid substitutions. In the case of p.Tyr16Asn and p.Arg180Thr, further support for a causative role arose from the observation that these mutations were recurrent in our cohort.
The majority of the new missense mutations occurred in amino acids conserved across multiple species with the exception of p.Arg180Thr. The bioinformatics analysis revealed that all of the new missense mutations have high predictive scores for pathologic effects according to the PolyPhen prediction software; although the SIFT software predicted that p.Phe679Ser, p.Trp1835Arg, and p.His2309Asp might be tolerant, a detailed description of these mutations is provided in Table 4. The putative causative role of the novel missense variants was analyzed based on the B domain-deleted crystallographic structure (Figure 1).
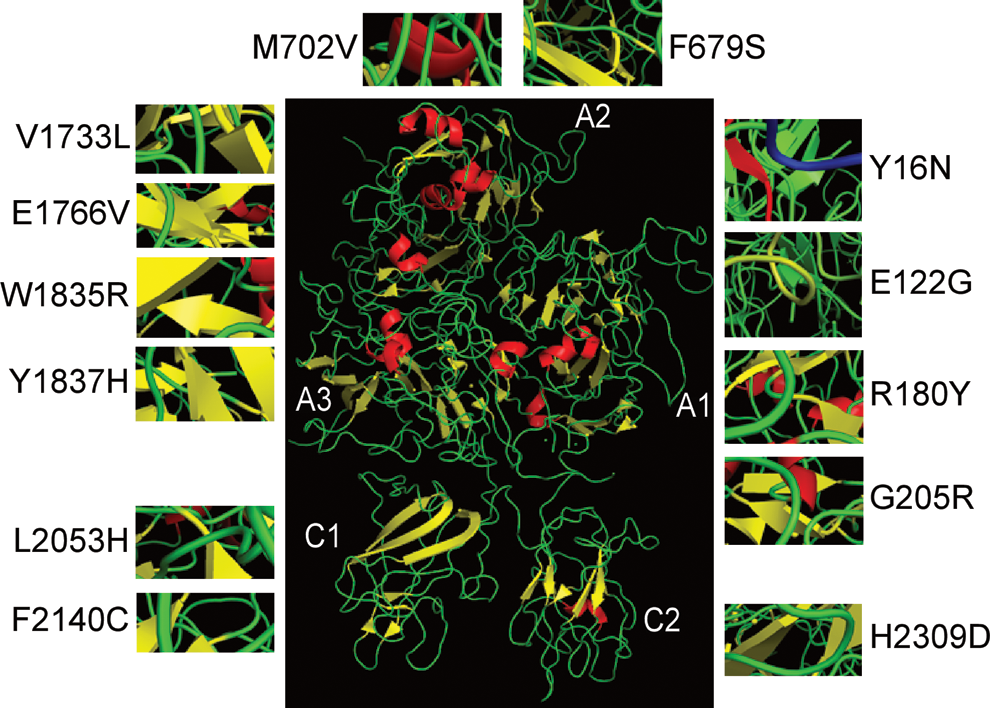
The crystal structure of the B domain-deleted factor VIII and the 3-dimensional structures of the novel missense mutations identified in our study.
Predicted Deleterious Effect Based on Multiple Alignment and Algorithms Studies.

Abbreviations: PolyPhen, polymorphism phenotyping; SIFT, sorting intolerant from tolerant.
aDegree of conservation among human (h), porcine (p), murine (m), and canine (c) factor VIII amino acid sequences.
A novel p.Glu122Gly missense mutation identified in 1 patient with mild HA was located within the Ca2+-binding site of the A1 domain 28 ; this mutation might alter the binding of calcium to FVIII and thus induce a structural change. The novel missense variation of p.Gly205Arg was detected in a patient with severe HA. This single-nucleotide substitution is located 1 base prior to the intron 5 donor splice site; therefore, it might alter the splice location. Met702 is located within the carboxy terminal of the A2 domain, an important region with respect to interactions with the FIXa protease domain. 29 A patient with severe HA was found to harbor the novel p.Met702Val missense mutation, which was predicted to modify the possible contact residues. Codons 1835 and 1837 are located near the FIXa-binding sites in the FVIII light chain 30 ; therefore, it is reasonable to assume that p.Trp1835Arg and p.Tyr1837His would cause suboptimal FIXa–FVIIIa complex assembly. The 4 novel missense mutations p.Arg180Thr, p.Glu1766Val, p.Leu2053His, and p.His2309Asp introduced structural instability by affecting the molecule’s electrostatic surface (Figure 2). The altered charge associated with p.His2309Asp might interfere with the interaction between FVIII and vWF. 31 At Phe2140, which is located in the C1 domain, the introduction of cysteine would potentially allow the formation of an extra disulfide bridge within FVIII, thus causing a major structural rearrangement. Aside from the above mutations, the structural or functional consequences of the 4 additional missense mutations (p.Tyr16Asn, p.Phe679Ser, p.Val1733Leu, and p.Glu1766Val) could not be deduced.
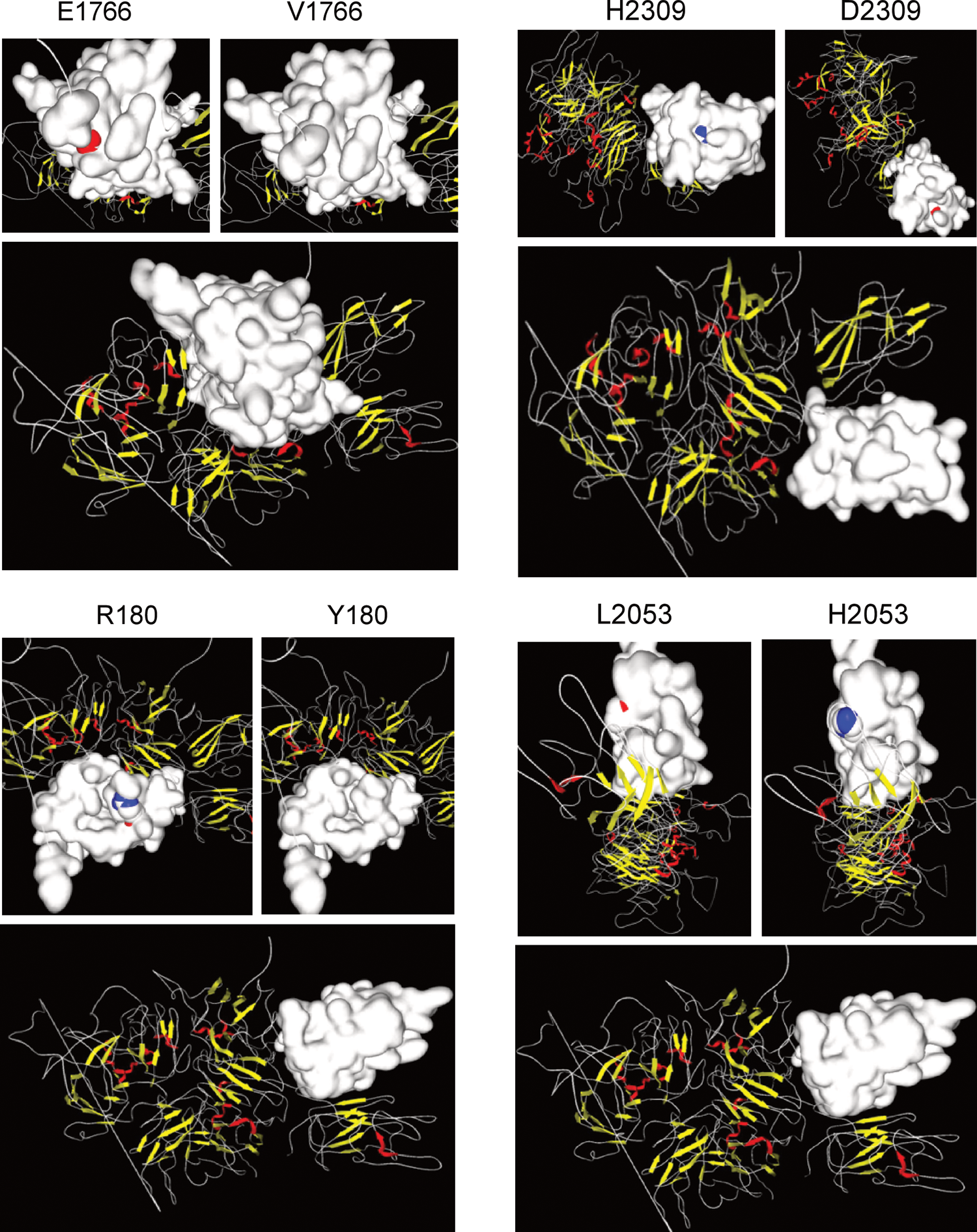
The effects of the 4 novel missense mutations (p.Arg180Thr, p.Glu1766Val, p.Leu2053His, and p.His2309Asp) on the factor VIII electrostatic surface (blue, positive charge; red, negative charge; white, neutral charge).
Inhibitor Development
Inhibitor development is a complex, multifactorial immune response involving both patient-specific and treatment-related factors. The F8 gene mutation type has been shown to be a significant risk factor for developing an inhibitor. 32 Twelve patients with severe HA in our study have FVIII inhibitor (Table 5). The prevalence of inhibitors in severe HA was 6.45% (12 of 186). The highest risk (40%) in our study was observed in Inv1 (2 of 5); however, due to the limited number of patients, the inhibitor risk was difficult to assess in this type of mutation. The OR of the nonsense mutations was found to be statistically significantly higher. Although the risk associated with a large deletion was greater, no statistically significant differences were identified. Among the 23 patients with severe HA with missense mutations and the 8 patients with splice-site mutations, none developed inhibitors. As previously described, null mutations with complete absence of protein are associated with a higher risk of inhibitor development than missense mutations that might permit some level of protein synthesis, given that the production of a nonfunctional FVIII protein could lead to a partial central tolerance to the altered FVIII protein. 33 Although the pathogenetic mechanism of Inv1 is similar to that of Inv22, 6 Inv1 conferred a higher inhibitor risk than did Inv22 in our cohort. This outcome might have resulted from the existence of an FVIIIB protein that was intracellularly encoded by the second gene transcript in the Inv22-positive patients. 34 Small deletions and insertions that affected the poly-A runs carried a lower inhibitor development risk (0 of 8) than did frameshift mutations occurring at other sites (2 of 20, 10%). The explanation for this phenomenon is that the partial restoration of the reading frame might have produced some FVIII molecules via polymerase enzyme slippage errors in the poly-A runs. 35 It seems that high-titer inhibitors would be more likely to present in nonsense mutation (3 of 6, 50%) and inversion (2 of 6, 33.3%). However, too few patients were available in the subgroups of high- and low-titer inhibitors to yield meaningful estimates.
Incidence and Odds Ratios of Inhibitor Development According to the F8 Genotype.
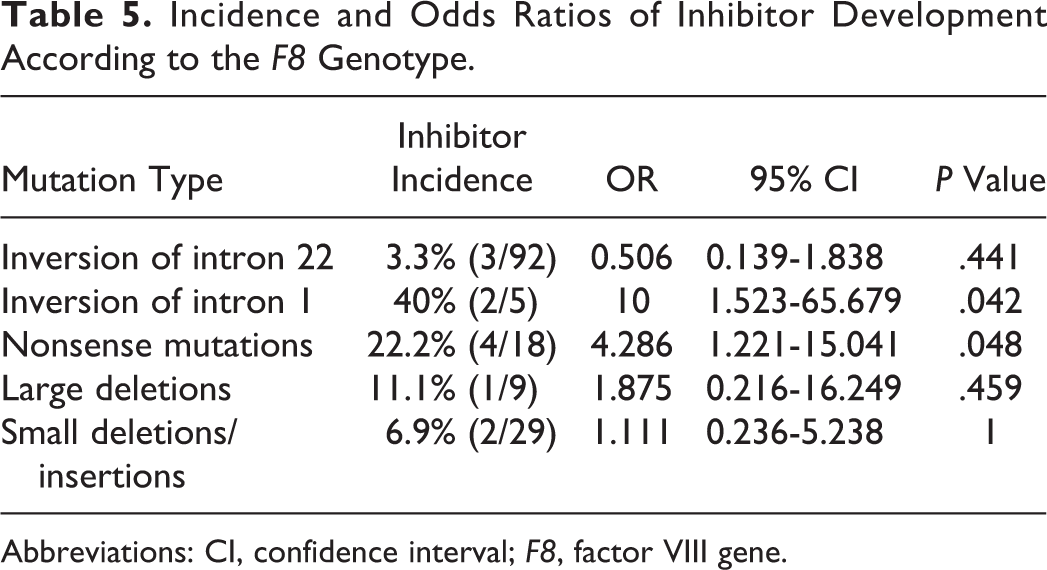
Abbreviations: CI, confidence interval; F8, factor VIII gene.
Conclusion
We reported on the spectrum of F8 mutations in the large series of HA in China. Our study revealed 42 of the identified mutations were novel, including 29 null mutations and 13 missense mutations for which the causality was demonstrated via bioinformatics. Aside from Inv22 and Inv1, we have disclosed several hot spots, including nonsense and missense mutations in the Arg sites and small deletions/insertions in the poly-A runs of the B domain. The genetic variations in the B domain were polymorphisms rather than causative mutations, and nonsense mutations were risk factors for inhibitor development.
Footnotes
Acknowledgments
The authors would like to thank Prof Man-Chiu Poon (Departments of Medicine, Pediatrics and Oncology, University of Calgary, Canada) and Prof Anthony Chan (Division of Pediatric Hematology/Oncology, McMaster Children’s Hospital, Canada) for critical review of the manuscript.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by research grants from the National Natural Science Foundation of China (81270587) and the Shanxi Scholarship Council of China (2009 key 7).