
Abstract
The story of factor X (FX) Friuli. Factor X Friuli was discovered in 1969 to 1970. However, the story of that disease was an international event since patients with this defect were studied in France and in Italy, and different diagnoses were reached—FVII; FX; combined prothrombin complex; and combined FII, FVII, and FX deficiencies. The diagnostic difficulties were due to the peculiar clotting pattern presented by these patients, namely, prolonged partial thromboplastin time, prolonged prothrombin time but normal Russell viper venom clotting time. Only suitable anti-FX antisera clarified the pattern. Altogether 12 homozygotes and 102 heterozygotes have been followed during 4 decades. Six homozygotes died, 2 of them due to HIV infection and 1 due to hepatitis B liver cirrhosis. The other 3 died of nontransfusion-related morbidity. Bleeding tendency has been moderate in agreement with the extrinsic or intrinsic system assay results—FX level of 4% to 5% is considered normal. Heterozygotes may present occasional bleeding manifestations usually during surgery or delivery. Molecular analysis have shown that the mutation responsible for the defect is a Pro343Ser substitution in exon 8. Chimeric FX Friuli mice have been useful in studying the effect of FX levels on embryonic or natal mortality of these animals. No new homozygote but several heterozygotes have been recently seen. The study of FX Friuli has revolutionized the diagnostic approach to FX deficiencies. The FX should be assayed by all assay systems. The FX Friuli has never been described in any other country, and all patients studied come from the Friuli Meduna River Valley.
Introduction
Congenital coagulation disorders have been discovered as a consequence of the description of a patient with a clotting pattern that did not suit the existing knowledge on blood coagulation at that time. There are only 2 exceptions to this rule, namely, factor VII (FVII) and FXIII deficiency. In both these cases, the existence of a congenital defect was postulated on the basis of laboratory clotting studies, which had allowed to surmise the existence of an unknown clotting factor, the absence of which could give rise to a new coagulation disorder. 1
The discovery of variants of all coagulation disorders, on the contrary, was always the result of investigations of a patient who was thought to have a known defect and turned out instead to present a different pattern from that expected as typical of the defect in question.
So hemophilia B was found because the patient showed a prolongation of thrombotest, namely, of a prothrombin time (PT) with an ox brain thromboplastin reagent. 2,3 The FX Friuli variant was first suspected because of the presence, in some patients, of a normal Russell viper venom clotting time (RVVCT) together with prolonged partial thromboplastin and PTs. 4 –7
The Complicated Story of the Discovery
Friuli, a northeastern Italian region, was in the past a poor area. The Meduna River Valley, where FX Friuli was discovered, was particularly depressed economically. The Valley was isolated to the north by a mountainous pass and to the south by narrow roads. Therefore, the population had few contacts with other communities. Intermarriages among known or unknown distant relatives was common practice as demonstrated by the fact that, even today, only about 20 last names are shared by about 80% of the population. These were and still are the geographic and socioeconomic conditions that fully explain the diffusion of autosomal recessive conditions as an FX defect. 6,7 Emigration was often the solution for survival. There were 2 types of emigration, namely, a transoceanic emigration (the United States, Australia, and Argentina) and a local European (Germany, Switzerland, Austro-Hungarian Empire, and France) emigration. The first was usually permanent and the latter usually seasonal. So it happened that 3 related patients from the Valley emigrated to France in the late 1940s or early 1950s, 1 in Troyes and Toulose and 2 in Paris. 8,9 The first patient was seen in a hospital of these first 2 cities and known as a “hemophilic.” Unfortunately, there are no records and the patient, after a few years, returned to Friuli and was subsequently investigated by us. The 2 patients living in Paris, when bleeding occurred, were referred to the Department of Hematology of the Hospital Hotel-Dieu, in Paris.
The 2 patients living in Paris were maternal cousins and were also related to the other patient. They were extensively investigated during several years. The results of those studies were 2 articles by Chevallier et al which was published in 1955 and 1959. 8,9
In the 1955 article, the patients were presented as cases of congenital hypoconvertinemia. 8 In the 1959 article, the same patients were considered as cases of FX deficiencies 9 (Table 1).
The Long Story of the Different Diagnoses Made During More Than a Decade for the Patients With FX Friuli.
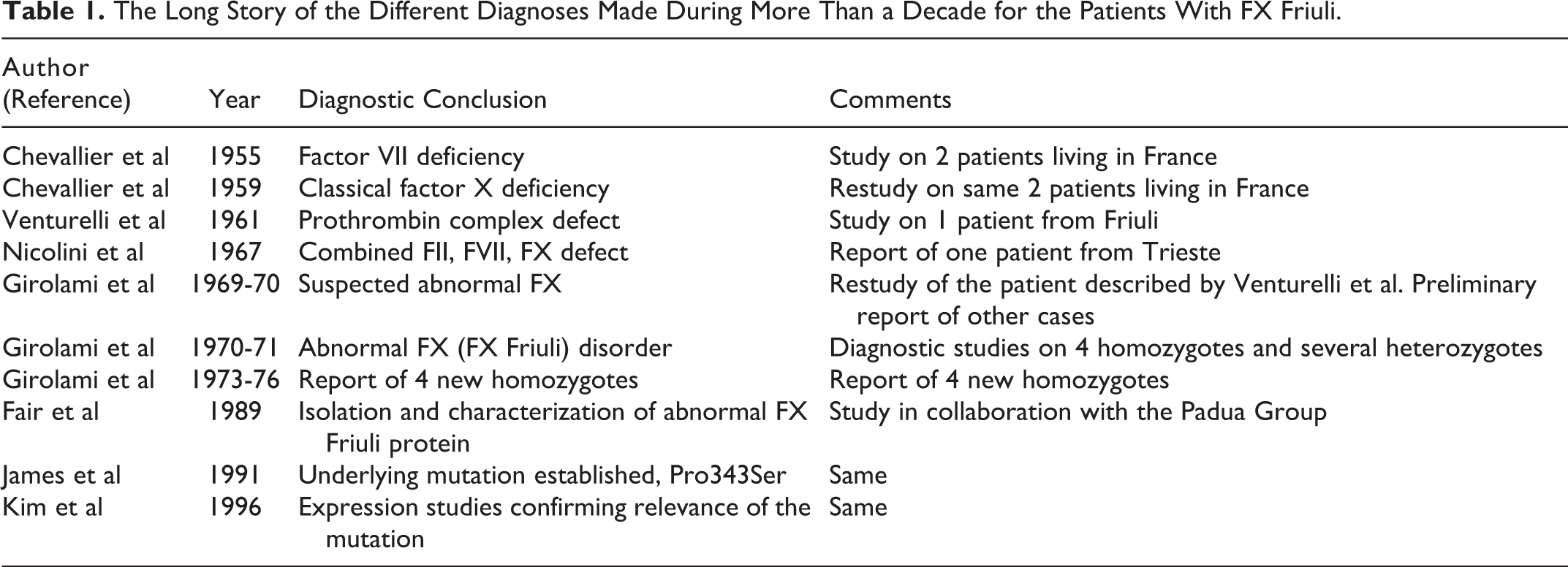
It is worth noting that Chevallier et al sent serum of 1 of the 2 patients to Prof Koller in Zurich for further study, and the conclusion was combined FVII and FX deficiency. 9
During the years 1956 to 1957, the distinction between FVII and FX had been firmly established by the description of 3 cases of FX deficiency in England, the United States, and Switzerland. 10 –13
In 1961, Venturelli et al who were responsible for the blood bank of the City Hospital in Udine, the capital of the Friuli region, described a patient who came from the same area as the Paris patients but was not related to them and concluded the diagnosis of prothrombotic complex defect. 14 In 1967, Nicolini of Trieste published an article in a local journal about a patient with a combined defect of FII, FVII, and FX 15 (Table 1). In early 1968, Dr Molaro, who had become responsible of the blood bank of the Pordenone City Hospital, always in Friuli, and had already studied the patient in Udine, got in touch with us in Padua. We then started a joint systematic study of the defect. Rumors about the existence of a bleeding tendency among the population of the Valley dated many decades ago. When our group started the study of the population, it was soon realized that memories and fear of bleeding were widespread. Many persons could recollect about relatives who had a tendency to bleed profusely after traumas or incidental cuts. Field work in the Valley allowed us to find several patients who presented the same peculiar defect.
In 1969, our group published the first article on the patient suspecting the existence of an abnormal FX. 4 In early 1970, we published in the Scand J Hematol, an article that was a restudy of the case previously published by Venturelli et al. 14 We again concluded on the suspicion of the presence of an abnormal FX. 5 In 1968 to 1969, this patient was working in Milan and was seen by Prof De Cataldo of Milan City Hospital, who sent a sample of plasma to Prof Denson in Oxford. At that time, Prof Denson had developed suitable antibodies and had described abnormal FIX variants and abnormal FX forms. 16,17 The patient listed as Dec in the table published by Denson et al 17 refers to the plasma he had received from Prof De Cataldo, namely, it belonged to a patient with FX Friuli. Prof Denson was then kind enough to carry out neutralization and immunodiffusion tests in a few new patients discovered in the meantime. Always in 1970, an article dealing with a large kindred was published in the British Journal of Haematology, and the diagnosis was firmly established. It was a new defect, due to the presence of an abnormal FX. 6 This was confirmed the following year with the report of another family which appeared in Blood. 7
Subsequently, we discovered other cases and established that all patients came or had ties with the original valley (the Meduna River Valley). 18 Several heterozygotes were also discovered who, contrary to heterozygotes for FVII deficiency, could occasionally have a mild bleeding tendency. 19
Studies and Achievements After the Discovery
After the original 3 families, which contained 5 homozygotes (3 patients in 1 family and 1 patient in each of the 2 other families), 7 additional homozygotes were found belonging to 7 other families. Six of these families all came from the same Valley, and distant relationship could often be traced with the index family cases. 20
Only 1 patient came from outside Friuli, from the city of Trieste. This patient, as stated above, had been reported in 1967 as a case of combined deficiency of FII, FVII, and FX. 15 Despite the fact that we could not trace any relation with the index family or the other families, however, due to the geographical vicinity and to the historic emigration to the city of Trieste from Friuli in search of work opportunities, it is plausible that the ancestors of this patient also came from the Meduna River Valley. 21
Characterization of the Protein, Description of the Genetic Abnormality, and Molecular Modeling
Protein isolation and purification studies and genetic studies were carried out in 1989 to 1995 in collaboration with Dr Fair and Dr James of the University of Texas at Tyler. It was shown that the heavy chain of FX Friuli was abnormal, whereas the light chain was identical to the wild type. 22 The defect was then found to be due to a Pro343Ser mutation in exon VIII. 23 In 1996, expression studies carried out in collaboration with Dr Kim et al of the same university confirmed the significance of the mutation. 24 In fact, recombinant FX Friuli was shown to behave in clotting and immunological systems as plasma FX Friuli. Crystallographic studies have allowed the identification of an extra H-bond in FXFr. Such H-bond is likely to cause a new side-chain bonding of the Ser343 residue with residue Thr318, which closely lies in front of it. The consequent structural changes of the molecule could justify the peculiar activation pattern of FX Friuli. 24 This long fascinating story about these patients is summarized in Table 1. The mutation at the basis of FX Friuli (Pro343Ser) has never been reported in other areas of Italy or of other nations. This is true for homozygotes or heterozygotes and even for compound heterozygotes. This indicates that the founder was local.
Homozygotes and Heterozygotes
Altogether 12 homozygotes were identified and studied. The last one was discovered in 1976. 25 Six patients died during these years (Table 2), whereas 6 are still alive and in fair condition (Table 3). 20
Causes of Death in the 6 Homozygote Patients Deceased During the Observation Period.a
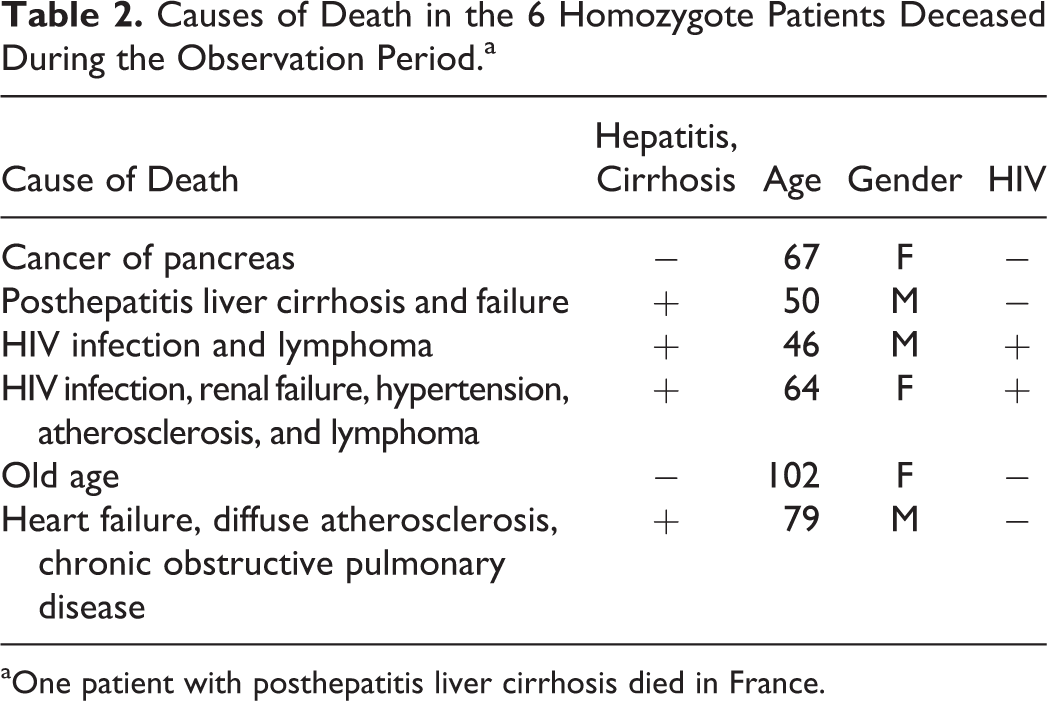
aOne patient with posthepatitis liver cirrhosis died in France.
Present Status of the 6 Homozygous Patients Still Alive.a
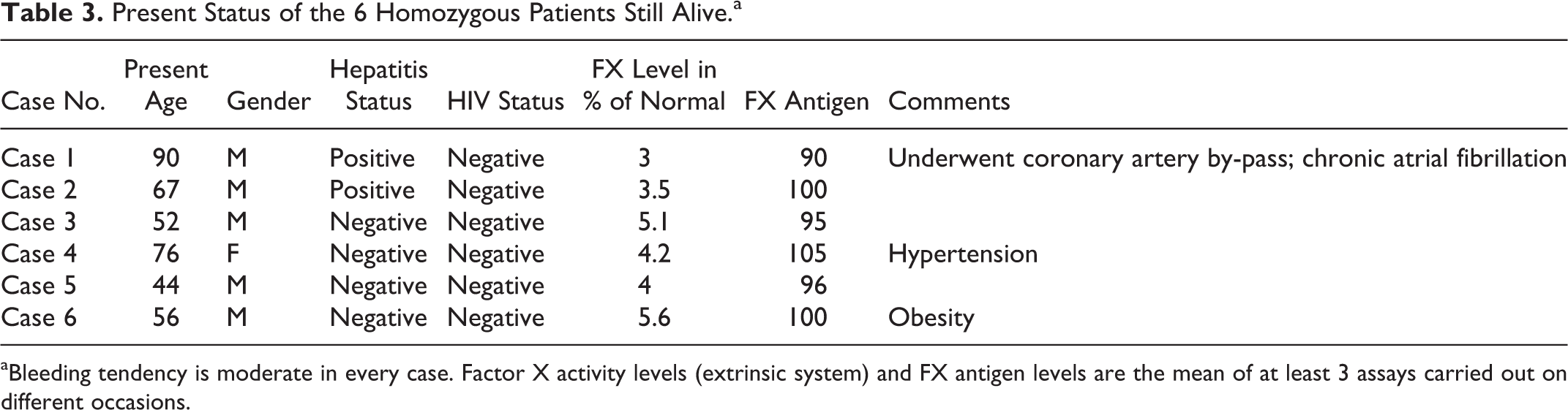
aBleeding tendency is moderate in every case. Factor X activity levels (extrinsic system) and FX antigen levels are the mean of at least 3 assays carried out on different occasions.
The most important fact was to study almost the entire population of the Valley (almost 2000 people) in search for heterozygotes. 19 By now, after almost 50 years, 102 patients met the criteria for heterozygosis. All homozygotes have to be considered as potential bleeders even though the severity of the bleeding manifestations is variable and usually moderate. 4 –7
No brain hemorrhage or hemarthrosis have ever been seen in FX Friuli patients. This is in contrast with classical FX deficiency in which these bleeding manifestations, particularly the first one, are common. 26,27 This indicates that FX levels of 3% to 6% of normal as seen in homozygote Friuli patients are sufficient to protect from such severe hemorrhagic symptoms. The normal FX activity levels in our laboratory are 85% to 115% of normal.
Heterozygotes, who have FX levels of 40% to 60% of normal, may present sometimes a mild bleeding tendency, especially after delivery or surgery. 19 This is a striking difference from heterozygotes of FVII deficiency who are usually asymptomatic. 28
Comorbidity of Patients With FX Friuli
During all these years, we have compared the nonhematologic mortality observed in the 12 homozygotes and 102 heterozygotes with FX Friuli with sex-matched, age (±5)-matched, unaffected family members. Both affected and their unaffected pairs were periodically checked every 12 to 18 months, for a mean observation period of 25.2 years (range: 2-39 years).
Symptomatic homozygous patients were checked in Padua or in local hospitals more frequently according to the need. The 3 homozygous surviving patients living outside Friuli (1 in Paris, 1 in Milan, and 1 in Trieste) are occasionally seen during their periodic visits in Friuli or Padua.
The comparison of the main morbid conditions which appeared in patients with FX Friuli and in the control group are gathered in Table 4. The number of death were 11 (6 among homozygotes and 5 among heterozygotes). In the control group, the number of death was 5. There were 4 deaths due to hepatitis or HIV infection in 4 homozygous patients, none in the control group.
Major Diseases and Fatalities Encountered in Patients With FX Friuli (12 Homozygotes and 102 Heterozygotes) and in the Control Group During the Long Observation Period (Mean: 25.2 Years).a

aAtherosclerosis and atherothrombosis include myocardial infarction (MI), other acute coronary syndromes, ischemic stroke, peripheral arteries diseases. Venous thrombosis include superficial and deep vein thrombosis.
The most common difference noted in the 2 group of patients was replacement therapy. As expected, it was present in 27 instances in the patient group and only in 1 patient in the control group (whole blood transfusion after delivery).
Arterial diseases (myocardial infarction, other acute coronary syndromes, ischemic stroke, peripheral arterial disease, and angina) occurred in 8 patients among the first group and in 7 among the controls (Table 4). Venous thrombosis was never seen in the patient group, but 3 patients of control group had venous thrombosis (2 after pregnancies, 1 after trauma). None of these patients showed Arterial thrombosis (AT), Protein, C and Protein S deficiencies, or FV Leiden or FII polymorphism.
Laboratory Implications
The description of FX Friuli had important implications in the diagnosis of prothrombin complex defects, mainly of FVII deficiency. 6,28,29 Till 1970, the criteria for the diagnosis of FX deficiency included prolonged partial thromboplastin time (aPTT), prolonged PT, and prolonged RVVCT. Those for FVII deficiency were prolonged PT but normal aPTT and RVVCT. The FX Friuli showed an intermediate pattern, namely, prolonged PTT and PT but normal RVVCT which created at the beginning a lot of confusion and led to the description of some patients as cases of FVII deficiency or of combined FVII and FX defect (Table 2). It is worth noting in this regard that the patient who became known as Mr Stuart had also been originally described as a case of hypoconvertinemia, namely, FVII deficiency. 30
The variability of the defect was subsequently confirmed by the description of variants defective only in the intrinsic system (FX Melbourne) or only in the extrinsic system (FX Padua). 29,31 –36 All these studies confirmed the great heterogeneity of the defect and indicated the need for multiple assay systems, extrinsic, intrinsic, and RVV dependent in the evaluation of suspected FX defects. The pattern is further complicated by the existence of congenital combined FX and FVII deficiency due to gross alterations in chromosome 13, where the genes controlling these 2 factors are closely related. 37,38
Pregnancies and Oral Contraceptives
During the observation period (1968-2010), there was 1 delivery among homozygotes. The event was accompanied by excessive bleeding that required replacement therapy. Four additional deliveries had occurred before the onset of the study, and they were also reported to have presented excessive bleeding with consequent substitution therapy. Among heterozygotes, 9 deliveries occurred during the observation period, and excessive bleeding was reported in only 2 of them. Eight additional deliveries had occurred before the start of the study; however, no sure information could be gathered about them.
None of the homozygotes took oral contraceptives (OCs) or hormonal replacement therapy. On the contrary, OCs were taken by 6 heterozygotes without any adverse effect.
Therapeutic Measures and Therapy-Related Diseases
All bleeding patients were treated on demand with plasma or several prothrombin complex concentration (PCC). 39 No immediate, untoward reaction was ever observed. However, hepatitis and HIV infection were seen in 4 and 2 patients, respectively. Two patients with hepatitis are still alive (Table 3).
Because the bleeding tendency was never severe or relapsing, no prophylaxis was ever attempted except in 1 patient for a short period of time. 40 No inhibitor ever appeared and this seems in agreement with those studies that indicate that cross reacting material (CRM+) patients, namely, those who have an abnormal, even though functionally defective, protein are less prone to see development of antibodies. 41
Tranexamic acid was often used, unless hematuria was present, with apparent good results. Two of the 12 homozygotes who might have needed anticoagulant therapy because of atherosclerosis and atrial fibrillation were kept off the drugs on the basis that their natural anticoagulation might be sufficient. However, both patients received low-molecular-weight heparin and acetyl salycilic acid (ASA) together with FX substitution therapy during arterial interventions. 40,41
The 2 patients living in France received several PCC and became infected with hepatitis B and C. One of the two patients died because of liver cirrhosis. The other is still alive at the age of 90. 42
Two patients developed HIV infection and both died at the age of 46 and 64, respectively. 20,43 No plasma exchange was ever carried out. None of the heterozygotes, including those who had received whole plasma replacement therapy and, in 2 instances, a single 500 U of PCC-developed hepatitis or HIV infection.
Mutations in the Area Close to the FX Friuli
Mutation in the area close (−6+6 codons) to the Friuli codon is rare (Table 5). 44 –47 The majority of them are represented by heterozygotes or compound heterozygotes. Only 2 cases (1 patient from Germany and 1 patient from Italy) are due to homozygosity for the same mutation (Cys390Phe). Therefore, few genotype–phenotype considerations are possible. However, it is worth noting that the Cys390Phe mutation is characterized by a type I defect in contrast with the type II defect of FX Friuli. 44,47
Main Features of FX Deficiencies Due to Mutations Close to the Pro343Ser (Now Pro383Ser) Mutation.a

aEponyms between parenthesis have been tentativelly allocated by the authors of the present article.
Note. n.r. = not reported.
Chimeric Studies on Murine FX Friuli
The moderate FX defect due to the Pro343Ser mutation, present in FX Friuli patients (4%-5% of normal), has been utilized to produce chimeric murine FX Friuli (FX F/F) to investigate the embrionic or perinatal mortality in comparison with Knockout FX animals (FX -/-). 48 It was shown that the FX levels seen in FX Friuli mice (FX F/F) with 4% to 5% FX activity were sufficient to eliminate natal lethality. Interestingly, even compound heterozygous animals (FX -/F) with FX levels of 1% to 3% showed no natal lethality. 48 Furthermore, it was shown that crossing hemophilia B mice with FX Friuli mice reduced or eliminated the thrombotic events and the increased mortality seen in hemophilia B mice with FVIIa overexpression. This was maintained to indicate that FX levels may play a role in the negative effects induced by FVIIa overexpression. 49
Conclusions
The existence of a bleeding tendency among the population of the Valley had been suspected for many years. It was reported by 1 tradition that some soldiers from the Valley, during the First and Second World Wars, had died because of excessive bleeding from combat-related wounds rather than because of the wounds themselves. We could not secure any confirmation of these stories, but we think they are likely to be true since all homozygotes observed by us during these years were often severe bleeders after trauma or surgery.
Since no other homozygous, heterozygous, or compound heterozygous patient with the same mutation has been discovered in other parts of the world, it has to be concluded that the founder was a local patient. In other words, the Pro343Ser or, according to more recent numbering, the Pro383Ser mutation has never been described outside the ethnic Friulian population. 50 –55
In 1983, we described a family with 5 heterozygotes patients from outside Friuli, in the Veneto Region, and thought it to be the expression of a separate mutation. Further family studies showed that an ancestor of this new family came from the original Valley of Friuli. 56
There are 2 mutations of FX geographically closed to the Friuli area, named FX Vorarlberg (Austria) 57 and FX Padua (Italy). 32 In both cases, the mutation is different and the clotting pattern is also different. Furthermore, no relation could be traced among these 3 areas.
No new case of homozygosis has been reported since 1976, the last case was diagnosed. 25 The last patient born with the clotting abnormality goes back to 1973. 18
Several heterozygotes have been described but the mobility of the population out of the Valley have reduced the chance of marriages between 2 heterozygotes.
Nowadays, you may find several heterozygotes outside the Valley and even outside Friuli or outside Italy, for example, in France. The 4 children of the 2 patients originally studied in Paris and found, as expected, to be heterozygotes, still live in France. In such country, they will have only an infinitesimal chance to mate with other heterozygotes, if any.
Another astonishing fact of this disease is the longevity demonstrated by these patients despite the clotting defect. None of the patients died because of bleeding. The deaths among the homozygotes were due to cancer in pancreas, HIV infection (two cases), and liver failure caused by hepatitis B cirrhosis (Table 2). One patient died at the age of 92. 20 One of the surviving patients is in fair conditions at the age of 90. 42 Atherosclerosis has been observed in 3 homozygotes and a few heterozygotes, but no venous thrombosis was ever observed. The autopsy findings in the index patient who died at the age of 70 revealed thickened arteries but no significant plaques. The patient was a moderate drinker and died of pancreatic cancer. 58
The lack of venous thrombosis in FX deficiency had already been noted by us in a search of the literature on all FX-deficient patients. 59 On the contrary, there is no apparent effect on atherosclerosis and this is in agreement with the interpretation that a clotting defect has little influence if any on arterial diseases. 60 –63 This is the only long-term study of a rare coagulation disorders.
The present study of these 12 homozygous and 102 heterozygotes patients with FX deficiency has cast some light on the effect of a moderate FX deficiency on survival and antithrombotic protection.
It is astonishing that the premature deaths were due to complications of the replacement therapy. For this reason and because spontaneous bleeding manifestations, even among homozygotes, are usually not severe, we think these patients should be treated on demand, especially on the occasion of surgery, delivery, or tooth extractions. Concentrates containing FXs, for example, the concentrate FIX + FX, are suitable. The safe hemostatic levels, in our experience, are around 45% to 50% of normal. This is in agreement with the observation that heterozygotes with FX activity levels around 40% of normal may bleed during surgery or tooth extractions. Once again congenital clotting disorders have supplied fertile ground for improving our knowledge of coagulation variants and the clotting mechanisms in general.
Footnotes
Acknowledgments
The authors wish to dedicate this article to the memory of Dr Brunetti and Dr Molaro who were coauthors of the original articles on FX Friuli which appeared in 1969 to 1971 and who are now deceased. The authors wish to thank the general practitioners of the towns of the Meduna River Valley where most patients with this peculiar coagulation disorder used to live and the staff of the Blood Transfusion Centers and of the Emergency Clinics of the local hospitals in Friuli. Without the dedicated and competent collaboration of all these persons, the study and the accumulation of the data concerning factor X Friuli could not have been possible.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported in part by the “Associazione Emofilia ed altre Coagulopatie delle Tre Venezie.”