
Abstract
Factor VIII (FVIII) inhibitors present major clinical challenge as a complication of hemophilia A in patients on treatment with FVIII concentrates and as acquired autoantibodies in patients without hemophilia A. We aimed to study the prevalence of FVIII inhibitors in Indian settings, risk factors involved in early development of inhibitors in patients with hemophilia, differences in their clinical behavior, and approach to treatment, in comparison to patients with acquired hemophilia. The overall prevalence of FVIII inhibitors in patients with severe hemophilia A was found to be 22.3%. Two cases of acquired hemophilia were reported. Due to heterogeneity of our study population, cases have been discussed individually. We observed that the early development of FVIII inhibitors in patients with hemophilia A is dependent upon an interplay of several risk factors that need to be studied in a multivariable analysis to bring out significant correlation with response to treatment. Also, they differ from patients without hemophilia A entirely in terms of presentation and management.
Keywords
Introduction
Inhibitors are antibodies that are usually targeted against either specific clotting factors, for example, factor VIII (FVIII), FIX, or phospholipids, that is, a lupus anticoagulant. Inhibitory alloantibodies to FVIII present a major clinical challenge as a complication of hemophilia A in patients on treatment with FVIII concentrates. 1 Many patient- and treatment-related risk factors have been identified which include age at first exposure to FVIII, family history, FVIII mutation status, type of FVIII product used, and on-demand versus prophylactic therapy. 2 The acquisition of FVIII inhibitors in the absence of hemophilia is a rare disease, which occurs mostly in adults. The common etiologies for the development of FVIII autoantibodies are autoimmune disorders, pregnancy, malignancies, and so on. 3 Hemorrhagic complications are more severe in patients with inhibitors, bleeding is more difficult to control, and patients have higher morbidity and mortality. With this study, we tried to find out the prevalence of FVIII inhibitors in the Indian settings, the various risk factors involved in the early development of inhibitors in patients with hemophilia A as well as the differences in their clinical behavior and approach to treatment, when compared to patients with acquired hemophilia.
Design and Methods
It was a retrospective study carried out in the Department of Hematology, All India Institute of Medical Sciences, New Delhi, during the period between January 2012 and December 2013. Patients with or without hemophilia A, who were found to develop FVIII inhibitors during the 2-year period, were enrolled in the study. Detailed history regarding clinical manifestations, frequency and severity of bleeding episodes, family history, age at first exposure to FVIII concentrates, and type of FVIII product used were compiled. Information was collected about the baseline investigations at the time of presentation including factor levels, mixing studies, and inhibitor titers by Bethesda assay and thereafter during follow-up visits at regular intervals. Also, the information regarding the treatment offered to these patients (post the development of FVIII inhibitors) was collected.
Results
Of 122 patients with hemophilia A, 67 (54.9%) patients had severe hemophilia A, apart from 50 (40.9%) and 5 (4.2%) cases, each of moderate and mild hemophilia A, respectively. Overall, the prevalence of FVIII inhibitors in patients with hemophilia A was found to be 13.1% (16 of 122). However, in patients with severe hemophilia, it was found to be 22.3% (15 of 67). Only 1 case of FVIII inhibitor was associated with moderate hemophilia A. During this period, we also observed 2 cases of acquired hemophilia.
Clinical picture of FVIII inhibitors in patients with hemophilia A was mainly dominated by hemarthroses and spontaneous hematomas (Table 1). Patients with hemophilia A were detected to have inhibitors either on routine testing or during periods of inadequate factor response. Patients with acquired hemophilia mainly presented with epistaxis, hematuria, joint pains, and purple ecchymotic patches over the face and trunk in the absence of any family history of similar episodes or exposure to factor concentrates.
Clinicohematologic Characteristics of Patients With FVIII Inhibitors.a
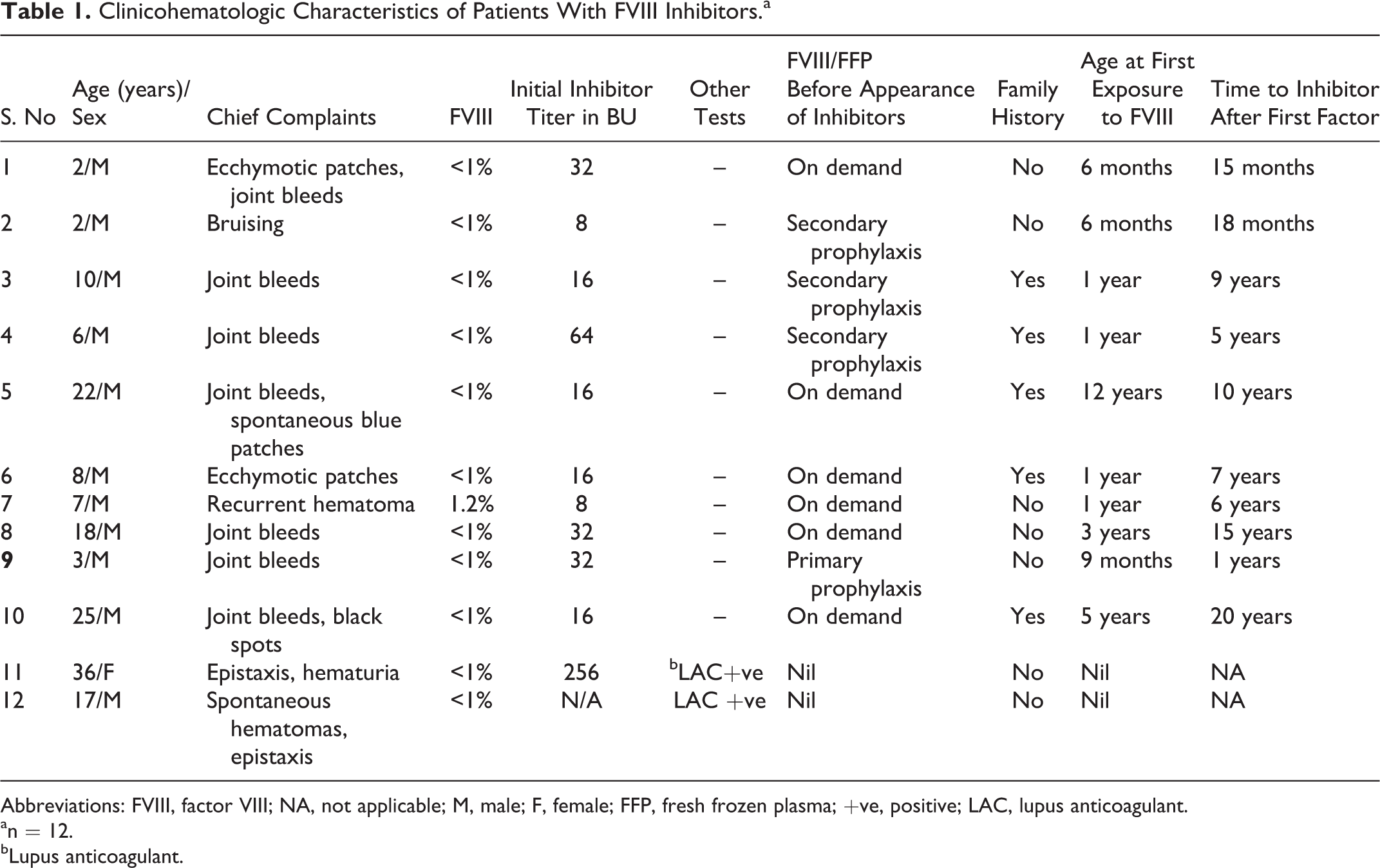
Abbreviations: FVIII, factor VIII; NA, not applicable; M, male; F, female; FFP, fresh frozen plasma; +ve, positive; LAC, lupus anticoagulant.
an = 12.
bLupus anticoagulant.
Majority (104 [85.2%] of 122) of our patients with hemophilia A registered during this period were receiving on-demand therapy with plasma-derived FVIII concentrates or fresh frozen plasma (FFP) during bleeding episodes. Only 18 (14.8%) of 122 patients were on primary or secondary prophylaxis. In all, 4 of 18 patients on prophylactic therapy and 12 of 104 patients receiving on-demand therapy developed FVIII inhibitors. Molecular analysis for the common mutations in patients with congenital hemophilia was not performed in our patients due to cost issues.
Among the hemophiliacs with inhibitors, the complete data and follow-up were available only in 10 of 16 patients as shown in Table 1. These patients have been studied in detail in terms of risk factors involved and the treatment strategies adopted before and after the development of inhibitors. At presentation, the median age of these patients was 7.5 years (range 2-25 years). A positive family history was found only in 50% (5 of 10) of patients. All of them were associated with high titer antibodies >5 Bethesda units (BUs) with median inhibitor titer of 16 BU and a titer range of 8 to 64 BU, prior to starting treatment for control of bleeding and eradication of inhibitors. Mean Bethesda titers in the prophylactic arm was 20 BU (range = 8-32 BU), and in the on-demand arm, it was 30 BU (range = 8-64 BU). The median number of exposure days before they developed inhibitors was 29 exposure days (range 10-100 exposure days). No inhibitors appeared after more than 100 exposure days in any of the patients. Owing to the heterogeneity of our patient population in terms of clinical behavior and their financial limitations, we have provided a brief case summary of these 10 patients.
Case 1
A 2-year-old male who received primary prophylaxis with FVIII concentrates (500 U/week) for 3 months before being shifted to on-demand prophylaxis due to cost issues. One year later, he was diagnosed with inhibitors (32 BU) during one of his bleeding episodes, when he showed no response to factor concentrates. He was started on recombinant activated FVII (rFVIIa) following which the inhibitor levels came down to 4 BU, 7 months later. He was later shifted to high doses of plasma-derived FVIII concentrates (1500-3000 U/week).
Case 2
A 2-year-old male who was initially receiving on-demand FFP transfusions for a year before being started on regular prophylaxis with recombinant FVIII (rFVIII; 250 U twice weekly). He developed inhibitors (8 BU) 6 months after starting prophylactic therapy. This inhibitor was transient and he no longer has any inhibitors. He was later shifted to on-demand therapy due to cost issues.
Cases 3 and 4
Brothers who received FFP as on-demand prophylaxis for 1 year before starting secondary prophylaxis with plasma-derived factor concentrates at a dose of 500 U twice weekly. They developed inhibitors at the same time (16 BU and 64 BU, respectively). Subsequently, they received only rFVIIa for bleeds and FVIII was discontinued. One brother had negative inhibitor screen a year later, whereas the other sibling had reduction in inhibitor levels (8 BU). They were restarted, after 16 and 7 months, respectively, on 50 units/kg plasma-derived FVIII, thrice weekly without any increase in inhibitor levels (or recurrence in the brother without inhibitor) at last follow-up.
Case 5
A 22-year-old male presented with a positive family history, who was receiving on-demand factor concentrates at a frequency of once or twice an year, before being diagnosed with inhibitors (16 BU), 10 years back. He was started on Novoseven (Novo Nordisk) (rFVIIa) during bleeding episodes. Later, he was shifted to immune tolerance induction (ITI) at a dose of 50 U/kg of FVIII concentrates thrice weekly, following which his inhibitor levels reduced to 4 BU. But, due to financial constraints, he is currently receiving on-demand therapy as and when bleeding occurs.
Cases 6, 7, 8, and 10
These cases received only on-demand prophylaxis—usually blood components, sometimes factor concentrate for bleeds (1-2 bleeding episodes a year with no joint deformity) and are on follow-up with regular testing for inhibitor titers (3 monthly).
Case 9
A case presented to us at age of 9 months with no prior family history. He was started on primary prophylaxis but developed inhibitors (32 BU) a year later. Subsequently, he received only rVIIa for bleeds, and prophylaxis was withheld. His inhibitors reduced to 16 BU after 7 months and he was started on rFVIII 50 units/kg thrice a week as immune therapy. His inhibitors increased to 32 BU. He had one subsequent gastrointestinal bleed for which he received rVIIa. But due to inadequate response, he was shifted to prophylaxis with FEIBA 50 units/kg thrice a week with no further bleeds. He has had no joint bleeds till date.
Among the patients without hemophilia A having inhibitors, the complete data and follow-up in 2 patients are described subsequently. Both cases were found in adults, with no sex predilection, in which etiology could not be defined.
Case 11
A 36-year-old female presented with history of ecchymotic patches on face and trunk and episodes of hematuria in the last 4 months. The patient had a very high inhibitor titer (256 BU). Factor VIII level was<1% and FIX, FX, and FII assays yielded normal results. Tests for lupus anticoagulant—kaolin clotting time and activated partial thromboplastin time–lupus anticoagulant—were positive but anticardiolipin antibodies and antidouble-stranded DNA antibodies were negative. She was started on prednisolone 15 mg along with cyclophosphamide 50 mg daily, following which her symptoms alleviated and inhibitor titers reduced to <5 BU within 6 months.
Case 12
A 16-year-old male presented with complaint of spontaneous hematomas, epistaxis, and joint pains for 4 months .No family history of bleeding was evident, both FVIII and FIX levels were less than 1%, and lupus anticoagulant was positive by 2 assays. Factor II, FX, and Bethesda titers could not be done in the patient. He was started on prednisolone 1 mg/kg daily following which he showed significant relief of symptoms with no fresh bouts of bleeding.
Discussion
In our study, the overall prevalence of FVIII inhibitors in patients with severe hemophilia A was found to be 22.3% which is similar to the prevalence rates found in Western countries 4 but is higher than other studies reported from India. 5 We found only 2 cases of acquired FVIII inhibitors in patients without hemophilia A over a period of 2 years which corroborates with the worldwide incidence of 0.2 to 1.0 case per 1 million persons per year. 6 Also, their clinical presentation was very different from that of patients with congenital hemophilia. Both the cases were associated with lupus anticoagulant. No cause could be defined for the difference in clinical manifestations between acquired and congenital hemophilia. 3 The relationship between the antiphospholipid antibodies and FVIII inhibitors is still controversial. They differ in terms of clinical presentation but are somewhat related, as both of them prolong the phospholipid-dependent coagulation tests. There have been arguments on the possibility that some patients with hemophilia may bear both types of anticoagulants, but this still remains an unresolved question because tests to detect LA without interference from the anti-FVIII inhibitors are lacking. 7
In the Indian settings, on-demand therapy with factor concentrates still remains the treatment of choice for patients with congenital hemophilia which is similar to the observation of other Indian published literature. 8 Most of our patients receive much less factor concentrates than the Western counterparts, that is, once or twice a year for bleeding episodes. Our study population was more heterogeneous with patients ranging from those receiving nil or on-demand prophylaxis to those on primary prophylaxis with rFVIII concentrates. We observed more number of inhibitor positive cases among patients receiving prophylactic therapy (22.2%) than those receiving on-demand therapy (11.5%), but heterogeneity of our patient population and smaller sample size precludes any such definitive conclusion. Also, development of inhibitors in patients with hemophilia A depends upon an interplay of various patient- and treatment-related risk factors such as FVIII gene mutations, family history, age at first exposure to factor concentrates, on-demand versus prophylaxis and the FVIII product type, intensity of treatment regimen, and so on; no single factor in isolation can be used in predicting early development of inhibitors. Studies aiming toward establishing correlation between treatment modalities with inhibitor development are interesting, but until the genetic factors regulating inhibitor development are clearly understood and can be used to properly subclassify patients, the association between the type of treatment and inhibitor formation will be hard to define. 9
In our study, all inhibitor positive patients had high titers >5 BU. No case of inhibitor titer <5 BU was observed, possibly because patients with low-titer inhibitors may not have a clinically relevant presentation and that the presence of inhibitors is only a transient event in them. 2 In patients with low-titer inhibitors presenting with minimal bleeding and who do not require surgery, it is usually appropriate to observe and monitor abnormal laboratory findings. If treatment is necessary, initial intervention can include routine hemostatic techniques and possible discontinuation of any antiplatelet or anticoagulation agents after assessment of comorbidities.
Also, the number of exposure days required for inhibitor development was in the similar range as in another Indian study. 5 Patients treated with FVIII preparations usually show a rise in the number of patients with inhibitors, particularly in the first 40 exposure days, after which the proportion of patients with inhibitors plateaus.
Family history of hemophilia was found in 50% of our cases which is similar to few other Western studies 2 which show a higher association (63%) with inhibitor development. However, the family history of inhibitors was seen in only 10% of our cases which is again similar to their results. 2 In our study, the earliest exposure to FVIII occurred at the age of 6 months with 70% of our patients receiving their first factor concentrate by the age of 1 year similar to other studies. 1,2 Also, the median age of presentation of patients with hemophilia having FVIII inhibitors was 7.5 years which is similar to most Western cohorts, which showed that majority of their patients also developed inhibitors by the first decade of life. 10 This may reinstate the fact that the prevalence of inhibitors in patients with hemophilia A is higher in the younger age-group, possibly due to acquisition of some nonsense mutations or large deletions, leading to significant molecular alterations and severity of bleeding manifestations, 1 but this needs to be confirmed by mutational analysis to establish a clear-cut correlation in our patients. Lack of information regarding the molecular mutations in patients with congenital hemophilia who later developed FVIII inhibitors was the drawback of our study, but ours being a developing country, most patients could not avail such expensive investigations.
Wherever feasible financially, patients were started on bypassing agents, that is, rFVIIa 90 μg/kg every 2 to 3 hours or FEIBA 50 to 100 IU/kg every 8 to 12 hours and continued till the bleeding stopped. They showed remarkable reduction or even disappearance of inhibitors. This implies that treatment should be started with bypassing agents as soon as diagnosis is made and at the lowest possible titer levels for improved treatment outcome. 5,11
Of 10 patients, 5 were further put on immune tolerance induction with low-dose regimens (50 units/kg) thrice or twice weekly depending upon the severity of disease. The ITI therapy is based on the repeated intravenous infusion of FVIII concentrates until the inhibitors are remarkably reduced and the recovery and half-life of FVIII are restored. Although several publications are reporting high success rate of about 70% inhibitor eradication following various immune tolerance induction protocols, they reflect heterogeneity of data and selection bias in terms of age, type of inhibitor response, follow-up of disease, and so on, which may affect the final outcome. 9,12,13
Among our patients without hemophilia A having FVIII inhibitors, the underlying etiology could not be defined in any of them. This is in concordance with the Western published data 14 according to which as many as 50% of cases remain idiopathic, despite thorough workup. Both the patients showed significant alleviation of symptoms along with reduction in inhibitor titers (256 BU to <5 BU in one of them) within few months of therapy with immunosuppressant drugs. This is in concurrence with the reported literature that advocates the administration of prednisolone at a dose of 1 mg/kg alone or in combination with cyclophosphamide 50 to 100 mg/d orally as soon as the diagnosis of acquired hemophilia is established. According to the published literature, poor prognostic features that may predispose to early relapse include older age at diagnosis, failure to achieve complete remission and malignant nature of underlying disease. All these risk factors were not present in any of our patients. According to the United Kingdom cohort, as many as 20% of patients relapse after a median gap of 7.5 months. 15,16 Thus, there is a need to follow-up these patients closely for reappearance of bleeding manifestations or sudden increase in inhibitor titers as soon as the drugs are stopped or their dose reduced.
Conclusions
The analysis of our data and the follow-up of the patients indicate that the most significant parameters in assessing response to treatment in patients with hemophilia developing FVIII inhibitors are as follows: (1) a low titer of inhibitor prior to treatment; (2) on-demand versus prophylactic therapy with FVIII; (3) age at first exposure to factor; (4) family history of inhibitors; and (5) time from inhibitor detection to start of eradication therapy. These variables have an important impact on the treatment costs as well. But no single risk factor in isolation can explain the correlation between early development of FVIII inhibitors and the response to treatment. This needs to be studied in a multivariable analysis to bring out statistically significant conclusions. Also, the differences in the pathophysiology of formation of allo- and autoantibodies to FVIII in patients with congenital and acquired hemophilia have a marked impact on the clinical presentation and treatment strategies in both groups of patients.
Footnotes
Authors’ Note
NS organized and analyzed the data, and drafted the article. RS and PM coordinated the study, participated in its design, and contributed to writing. HP, ST, MM, and TS provided valuable clinical inputs and reviewed the article. All authors read and approved the final article.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.