
Abstract
The aim of this study was to assess platelet reactivity in patients after ischemic stroke and to investigate the influence of hyperlipidemia (HL) on platelet activity markers. A total of 41 patients after ischemic stroke were divided into the following 2 groups: patients with HL and patients with normolipidemia. Expression of CD42b on resting, thrombin-activated blood platelets, and fibrinogen level was assessed. The CD42b-positive platelets were analyzed using the flow cytometer, anti-CD61, and anti-CD42b monoclonal antibodies. The results confirmed increased platelet reactivity to thrombin in all patients after ischemic stroke manifested by significantly lower CD42b expression and percentage of CD42b(+) platelets after activation by thrombin. The influence of HL on the expression of CD42b on resting and thrombin-activated platelets was not found. However, increased level of fibrinogen but no influence of HL on fibrinogen concentration was observed in patients after ischemic stroke. Increased susceptibility to platelet agonists was found in patients after ischemic stroke in the convalescent phase.
Introduction
Platelets play a crucial role in thromboembolism, and platelet activation and hyperreactivity contribute significantly to cerebrovascular events. Understanding mechanisms involved in platelet activation is important for the improvement of stroke prevention. Many studies suggested that platelets were excessively activated or hyperreactive in the acute and convalescent phases after cerebral ischemia. 1 –3 Estimation of various flow cytometry markers (increased expression of surface P-selectin and higher internalization of glycoprotein [GP] Ib–IX complex) reflects platelet activation and their functional response to agonists (reactivity). Platelet adhesion to subendothelial structures upon injury to a vessel wall is one of the first steps in the development of an arterial thrombus. Binding of the platelet receptor GPIb to subendothelial von Willebrand factor (vWF) plays a critical role in adhesion of platelets to the damaged vessel wall, at least in smaller vessels or stenosed arteries where shear stress is high. 4 The adhesion process is mediated by an interaction between the GP Ib–V–IX complex on the platelet surface and vWF, associated with collagen on the subendothelial surface. After initial adhesion, platelets are activated again and adhere to each other. This process leads to platelet plug formation or development of thrombus. 5
Not only enhanced platelet activation but also increased fibrinogen level plays an essential role in thromboembolic events. Fibrinogen is synthesized and assembled in hepatocytes and fibroblasts, then secreted into the plasma pool. 6 The plasma half-life ranges from 3 to 4 days. Pathogenic role of fibrinogen is suggested not only in acute thrombotic complications but also in the stable form of vascular diseases. 7
Pathogenesis of brain ischemia involves not only thrombotic but also immunoinflammatory mechanisms. Several studies showed that proinflammatory cytokine (interleukin [IL] 6 and tumor necrosis factor α [TNF-α]) and vWF plasma level are predictor factors for ischemic stroke incidence. Moreover, increase in proinflammatory cytokines (IL-1β, IL-6, and TNF-α) is significantly associated with acute stroke occurence. 8,9
One of the most important factors responsible for atherothrombotic events are low-density lipoproteins (LDLs). 10,11 Platelets activated by native and oxidized LDLs release platelet-derived growth factor, which increases the number of LDL receptors on macrophages. This reaction leads to augmentation of LDL accumulation in these cells and is responsible for atherosclerosis intensification. 12 Low-density lipoprotein also enhances platelet activation and reactivity. They cause intraplatelet acidification by Na+/H+ exchange inhibition leading to increase in susceptibility to platelet agonists. 13 Relationship between platelets reactivity and hyperlipidemia (HL) may be responsible for intensification of atherothrombotic events.
The aim of this study was to assess platelet reactivity in a group of patients after ischemic stroke and to investigate the influence of HL on platelet activity markers. These relationships may be an important target for ischemic stroke prevention.
Materials and Methods
The study group consisted of 41 patients after ischemic stroke at least 3 months prior to the investigation. The clinical diagnosis of ischemic stroke was based on medical history, neurological examination, and cranial computed tomography. The patients with stroke were divided into the following 2 groups: patients with normolipidemia (NL) and patients with hyerlipidemia; 20 patients with NL: LDL <130 mg/dL (3.36 mmol/L), mean 2.75 ± 0.5 mmol/L; total cholesterol (TCh) <200 mg/dL (5.17 mmol/L), mean 4.86 ± 0.55 mmol/L; 11 males, 9 females, mean age 69.8 ± 10.5 years; and 21 patients with HL: LDL ≥ 130 mg/dL (3.36 mmol/L), mean 5.27 ± 0.66 mmol/L; TCh ≥ 200 mg/dL (5.17 mmol/L), mean 7.48 ± 0.84 mmol/L; 12 males, 9 females, mean age 62 ± 10.2 years.
Twenty age- and sex-matched control subjects (CSs) were recruited from patients with discopathy or tension-type headache and no symptomatic cerebrovascular disease. The study participants were the patients of the Department of Neurology and Strokes, Medical University of Lodz, Poland.
The risk factors for ischemic stroke (arterial hypertension, ischemic heart disease, and body mass index) were similar in the study groups and the controls. All patients received aspirin (75 mg/d) as a secondary stroke prophylaxis and antihypertensive drugs (angiotensin-converting enzyme inhibitors, calcium channel blockers, and β-blockers). The inflammation marker, C-reactive protein, in study groups was within normal limits. Exclusion criteria for all study participants were diabetes mellitus, cancer, systemic and chronic inflammatory diseases, hemorrhagic diathesis, severe liver disease, renal failure, and anticoagulant treatment. To avoid the influence of stroke subtype on the results, the patients with lacunar and cardiogenic ischemic stroke were also excluded from the study.
Complete blood cell count, TCh, LDL, and high-density lipoprotein (HDL) levels were done in fasting state in all patients. The levels of TCh and HDL were estimated enzymatically with Olympus AU 6400 analyzer (ABS, Dallas, USA) and LDL level by the Friedwald formula.
The flow cytometry (FACScan; Becton Dickinson, San Jose, California) was used to estimate the expression of GPIbα (CD42b) on resting and thrombin-activated blood platelets. Blood from each patient was withdrawn without stasis to avoid activation of circulating platelets. The blood was collected into 2 tubes. The first tube contained 0.1 mL of blood and 1 mL 0.5% solution of paraformaldehyde in phosphate-buffered saline—the sample reflected platelet activity ex vivo; the other tube containing 0.5 mL of blood and 0.5 mL of EDTA was used to assess platelet reactivity after treatment with bovine thrombin. All platelet measurements were performed within 90 minutes after blood withdrawal. A fluorescein isothiocyanate-conjugated antibody to GPIIIa (anti-CD61-FITC; DAKO) was used as an activation-independent marker. Anti-CD42b-PE (DAKO:Glostrup, Denmark) antibody conjugated with phycoerythrin was used to assess platelet expression of GPIbα. To assess the extent of the nonspecific association of antibody protein with platelets, a control tube containing anti-CD61-FITC and nonfractionated PE-conjugated immunoglobulin G (Becton Dickinson) was used for each blood sample. To assess platelet ability to be activated by an agonist, the other sample was treated with 0.08 U bovine thrombin for 4 minutes. The reaction mixture was incubated at room temperature for 30 minutes in a dark room. Then the antibody-bound platelets were fixed with 200 μL FACSflow (sheath fluid, BD-Bioscience) liquid and analyzed.
Platelets were subtracted from other blood cells and identified by flow cytometry based on size and platelet-specific CD61 surface expression. Data were presented as the percentage of CD42b platelet expression and median of fluorescence intensity reflecting density of CD42b on platelet surface. WinMDI 2.8 was used to analyze the data collected with flow cytometry.
Data were analyzed by standard statistical analysis, Student t test, Shapiro-Wilk test, and Levene test. Before the analysis, all variables were logarithmically transformed to approximate the normal distribution. Data were expressed as mean value with standard deviation. Statistical analysis was performed using SPSS PC 11.5 and STATISTICA 6.0. P < .05 was considered statistically significant. The study was approved by the Ethics Committee of Medical University of Lodz, Poland (No. RNN/205/04/KB).
Results
The obtained results showed increased platelet reactivity to thrombin in all patients after ischemic stroke. After platelet stimulation by thrombin, the CD42b expression (HL 23.4 ± 10.5 vs CS 40.0 ± 8.9, P <. 001; NL 28.9 ± 13.5 vs CS 40.0 ± 8.9, P < .005) and the percentage of platelets CD42b+ (HL 19.5 ± 9.6 vs CS 31.5 ± 9.3, P < .001; NL 24.1 ± 12.7 vs CS 31.5 ± 9.3, P < .05) were significantly lower in both groups of patients with stroke compared with the controls (Figure 1A and B). No influence of HL was found on the CD42b expression after platelet activation by thrombin. There were no significant differences in the median of fluorescence CD42b (HL 23.4 ± 10.5 vs NL 28.9 ± 13.5, P = .155; Figure 2) and percentage of CD42b+ platelets after thrombin activation (HL 19.5 ± 9.6 vs NL 24.1 ± 12.7, P = .196) between HL and NL groups (Figure 1A and B).
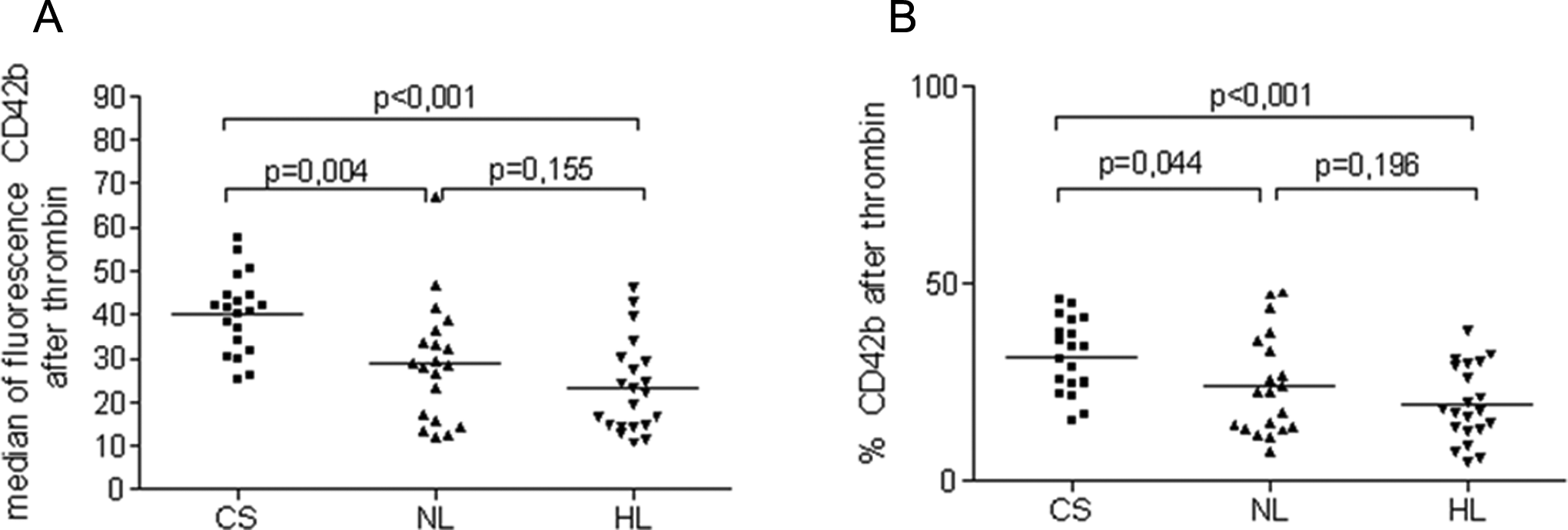
Median of fluorescence (A) and percentage of CD42b+ (B) in the study groups after thrombin activation. CS indicates control subject; NL, normolipidemic group; HL, hyperlipidemic group.
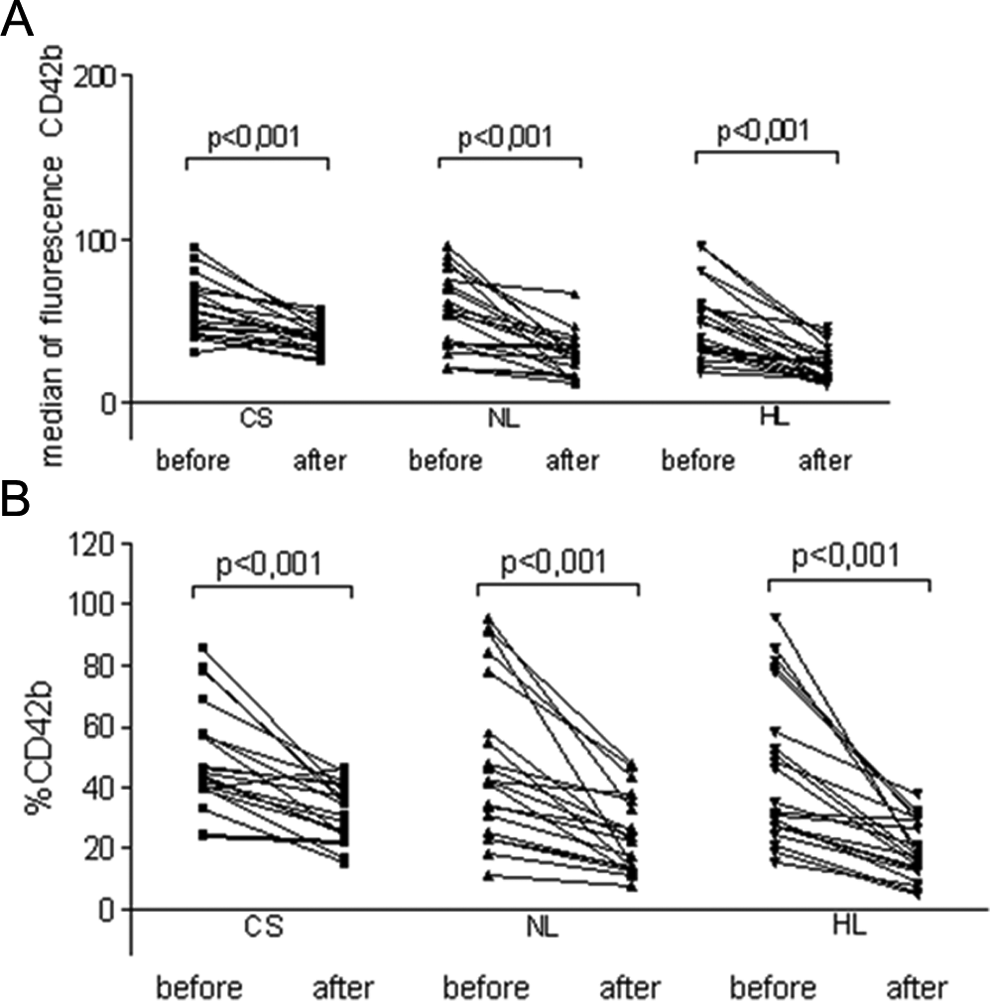
Median of fluorescence (A) and percentage of CD42b+ (B) in the study groups after and before thrombin activation. CS indicates control subject; NL, normolipidemic group; HL, hyperlipidemic group.
Assessing expression of CD42b on resting platelets, enhanced platelet activity was not found in stroke patients with stroke compared with the controls. There were no significant differences in platelet expression of CD42b before thrombin activation between stroke groups and the controls (median of fluorescence: HL 50.0 ± 22.9 vs CS 57.2 ± 17.1, P = .26; NL 54.8 ± 24.0 vs CS 57.2 ± 17.1, P = .73 and percentage of CD42b+: NL 48.7 ± 26.4 vs CS 49.6 ± 17.2, P = .9; HL 46.1 ± 24.8 vs CS 49.6 ± 17.2, P = .56; Figure 3A and B). Significant increase in fibrinogen level was found in patients after ischemic stroke compared with the controls, however, with no influence of HL on fibrinogen concentration (HL 442.7 ± 109.3 vs CS 294.4 ± 70.2, P < .001; NL 420.1 ± 70.0 vs CS 294.4 ± 70.2, P < .001 Figure 4) .
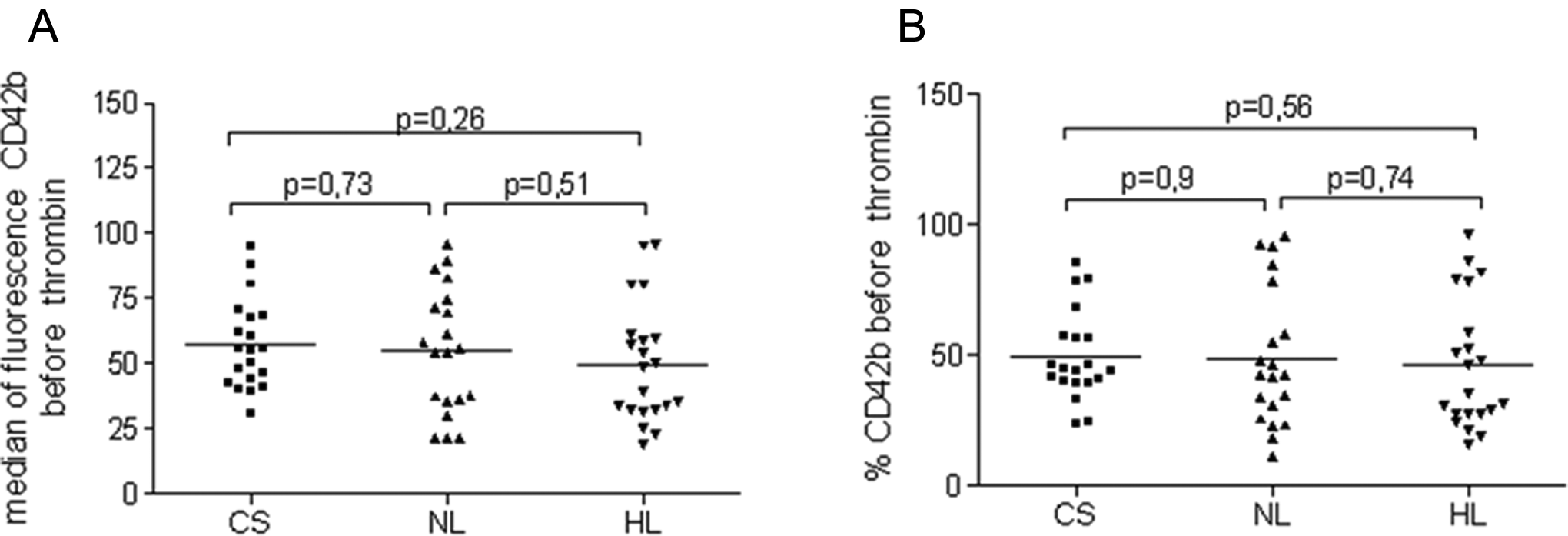
Median of fluorescence (A) and percentage of CD42b+ (B) in the study groups before thrombin activation. CS indicates control subject; NL, normolipidemic group; HL, hyperlipidemic group.
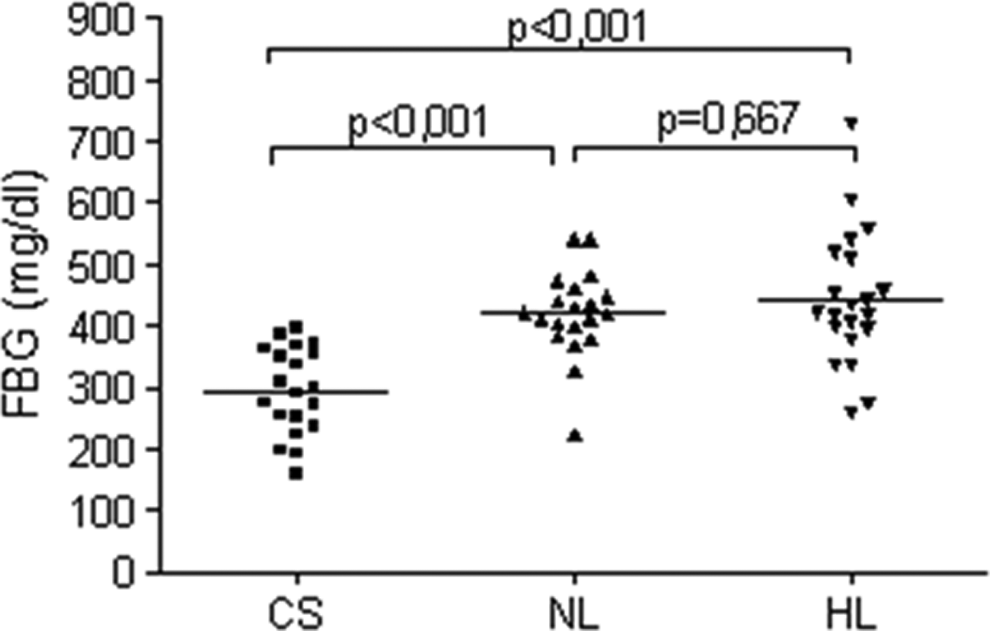
Fibrinogen levels in the study groups. CS indicates control subject; NL, normolipidemic group; HL, hyperlipidemic group.
Discussion
Despite antiplatelet treatment, platelet activation was found during acute and chronic phases of ischemic stroke 1 increasing the risk of secondary cerebrovascular events. Antiplatelet drugs may not prevent shear-induced platelet activation mediated by GPIbα and vWf. The GPIbα is an important molecule for platelet adhesion to damaged vessel wall and binds vWf under conditions of high shear stress. 14 The GPIb–IX–V complex not only mediates platelet adhesion but also transmits signals leading to platelet activation, aggregation, and secretion. 15 Some studies confirmed the correlation between GPIbα polymorphism, increased expression of GPIbα, and higher risk of ischemic stroke, 16,17 whereas other did not. 18
The obtained results did not show differences in the expression of GPIbα on resting platelets between the patients after ischemic stroke and the control group. These findings may indicate downregulation of platelets activity in the convalescent phase after ischemic stroke. However, platelets in patients after cerebral ischemia are more susceptible to the platelet agonist compared with the CSs. It was reflected by significant decrease in platelet CD42b expression (caused by GPIbα internalization) after stimulation by thrombin in both groups of patients with stroke. Increased susceptibility to platelet agonist may be responsible for atherothrombotic intensification and may be one of the mechanisms leading to ischemic stroke. It was shown in an animal model of stroke that VWF–GPIbα interactions are instrumental in thrombus formation after transient middle cerebral artery occlusion 19 and, what is interesting, complete blockade of GPIbα led to significant reduction in infarct volumes. 20 Thus, increased platelet reactivity may be a risk factor for another ischemic stroke, and inhibition of VWF–GPIbα interaction could be a promising target for stroke treatment.
Hyperlipidemia is a well-established risk factor for atherothrombotic events, in which platelet activation plays a significant role. Study of Stokes et al 21 showed that short-lasting (≤2 weeks) hypercholesterolemia induces proinflammatory and prothrombotic state in microcirculation. Hypercholesterolemia appears to convert the normal anti-inflammatory phenotype of microcirculation to a prothrombotic phenotype 22 ; however, the obtained results did not confirm the data. The influence of HL on the expression of GPIbα was not found in our study, it may be due to including the patients with large vessel diseases and excluding those with lacunar infarct. Thus, our results do not deny the significant influence of HL on the increasing risk of ischemic stroke. This association is additionally emphasized by lower mean age of patients with hyperlipidemic stroke compared to normolipidemic ones. It seems that GPIbα is not as adequate as sP-selectin concentration marker for the assessment of HL influence on platelet activity. 23
Fibrinogen is another factor related to atherosclerosis development. Elevated level of fibrinogen was found in patients with coronary heart disease, peripheral vascular disease, and carotid stenosis. 24 –26 There are also a few studies showing the positive relationship of fibrinogen concentration with ischemic stroke. 27,28 The cohort study of Rothwell et al 29 showed that the risk of recurrent ischemic stroke and acute coronary events in patients with previous cerebrovascular diseases (transient ischemic attack or stroke) increases linearly with fibrinogen concentration. The authors also observed a trend toward a stronger relationship between fibrinogen concentration and risk of acute ischemic stroke in patients with nonlacunar than lacunar stroke. The results of our study are concordant with Rothwell et al findings. 29 We also found a significant increase in fibrinogen level in patients after ischemic nonlacunar stroke. Observations of many authors as well as ours indicate that elevated level of fibrinogen may induce the prothrombotic state and may be one of the factors increasing the risk of recurrent vascular ischemia. High fibrinogen concentration contributes to atherosclerosis intensification not only by the increase in platelets aggregation but also by the enhancement of fibrinogen content in thrombus as well as change in thrombus structure, which leads to decrease in clot susceptibility to plasmin action. Influence of HL on fibrinogen level was not found. Enhancement of fibrinogen level was independent from lipid concentration.
In conclusion, increased susceptibility to platelet agonists was found in patients after ischemic stroke in the convalescent phase. Elevated platelet reactivity and increased level of fibrinogen may be responsible for atherothrombotic intensification and may be one of the mechanisms leading to a higher risk of ischemic stroke. Thus, patients after ischemic stroke require better prevention treatment, and inhibition of VWF–GPIbα interactions seems to be a promising target for this treatment.
Footnotes
Acknowledgments
We would like to thank Anna Augustyniak for her technical assistance.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The study was supported by a grant from the Medical University of Lodz, Poland (grant no. 502-15-385 and grant no. 502-03/5-062-01/502-54-009).