
Abstract
A family with a novel c.717_del frameshift and a c.3655C > T missense mutation of a disintegrin and metalloproteinase with thrombospondin type I motif, member 13 protein (ADAMTS13) is described. Family members have been under observation for 44 years. Two double heterozygotes have severe early-onset Upshaw-Schulman syndrome and require prophylactic plasma infusions. Analysis reveals that 2 weekly plasma infusions are not sufficient in preventing laboratory evidence of a thrombotic thrombocytopenic purpura (TTP) attack. Both the double heterozygotes also have a heterozygous factor V Leiden G1291A mutation. One underwent splenectomy, which did not reduce the frequency of TTP episodes but resulted in a recurrent pulmonary embolism and has necessitated lifelong anticoagulant therapy. The other has mild chronic renal failure and has had episodes of atrial fibrillation and cerebral infarction. Of the 3 heterozygotes in the family, 1 has had episodes of mild thrombocytopenia.
Keywords
Introduction
Thrombotic thrombocytopenic purpura (TTP) is a microangiopathic disorder with a high mortality rate if untreated. It is due to a deficiency of the a disintegrin and metalloproteinase with thrombospondin type I motif, member 13 protein (ADAMTS13), which cleaves Von Willebrand factor (VWF) between Tyr 1605 and Met 1606. As a result of this deficiency, unusually large VWF multimers are present, which are more efficient in binding to platelets causing their aggregation and resulting in the clinical and laboratory features of TTP. Most cases of TTP are acquired and are caused by antibodies acting as inhibitors of the ADAMTS13 protein. These are treated by measures to remove the antibody by plasmapheresis and plasma replacement using fresh frozen plasma (FFP). Immune therapy with rituximab is also used. In rare families, there is a congenital deficiency of ADAMTS13 causing recurrent TTP, which can be corrected by plasma infusion. This condition was first recognized in the cases described by Upshaw 1 and Schulman 2 and is known as the Upshaw-Schulman syndrome (USS).
Schulman first described a girl with the syndrome in 1960 and updated the report in 1967. 3 This patient with severe thrombocytopenia repeatedly had a striking rise in the platelet count after the administration of fresh plasma or whole blood. Eventually, she was maintained on FFP every 2 weeks.
The case reported by Upshaw in 1978 was a 29-year-old female who had had attacks from the age of 6 months which were later recognized to be due to TTP causing thrombocytopenia and microangiopathic hemolysis. Between the ages of 18 and 29, 32 episodes occurred. They occurred spontaneously or following infection, surgery, pregnancy, and pancreatitis. Partial “remission” after infusion of packed cells and a marked response after infusion of whole blood or plasma suggested deficiency of a plasma factor to which the thrombocytopenia and anemia were responsive. She was subsequently managed by plasma infusions.
Homozygous or compound heterozygous mutations of the ADAMTS13 gene 4 on chromosome 9q34 may cause a secretion defect and in some cases a mutant protein with loss of enzymatic activity and decreased binding to substrate. 5 Over 140 mutations have been found so far. 6 These can cause an early-onset or a late-onset phenotype. 7 Some cases present with severe neonatal jaundice. Clinical signs may be mild in children and later become more apparent with trigger factors such as infection, trauma, alcohol abuse, or pregnancy. Other cases may not be diagnosed until later in life. One case is reported as presenting at the age of 63 years with no history. 7 A phenotype–genotype correlation has been suggested. 8,9
These patients have no antibodies to ADAMTS13 and are treated by replacement therapy when symptomatic which promptly averts an attack. They do not require plasma exchange. They can go for long periods without clinical features. Trigger factors may work by increasing VWF production and altering the balance between it and the minute amounts of ADAMTS13 in their plasma.
Some patients, however, have frequent severe attacks and need prophylactic plasma infusions to prevent these.
It is estimated that there are about 150 patients alive in the world today with this rare and fascinating disease. 6 Two of patients from 1 family have been under care in our institution for a period of 44 years. This is the longest follow-up of patients exceeding a 24-year report of a patient in 2004. 10 This review illustrates particular problems that were accounted and the measures taken to treat them.
Materials and Methods
Clinical details and routine laboratory data are described in the family description.
Measurement of ADAMTS13 and Molecular Analysis
Citrated plasma samples were evaluated for ADAMTS13 activity and functional ADAMTS13 inhibitors at the University Clinic for Hematology and Central Hematology Laboratory, Bern University Hospital, and the University of Bern, Switzerland.
Levels of ADAMTS13 were kindly initially measured by M. Furlan in 2000 using a immunoassay and a collagen-binding assay and are shown in Figure 1.
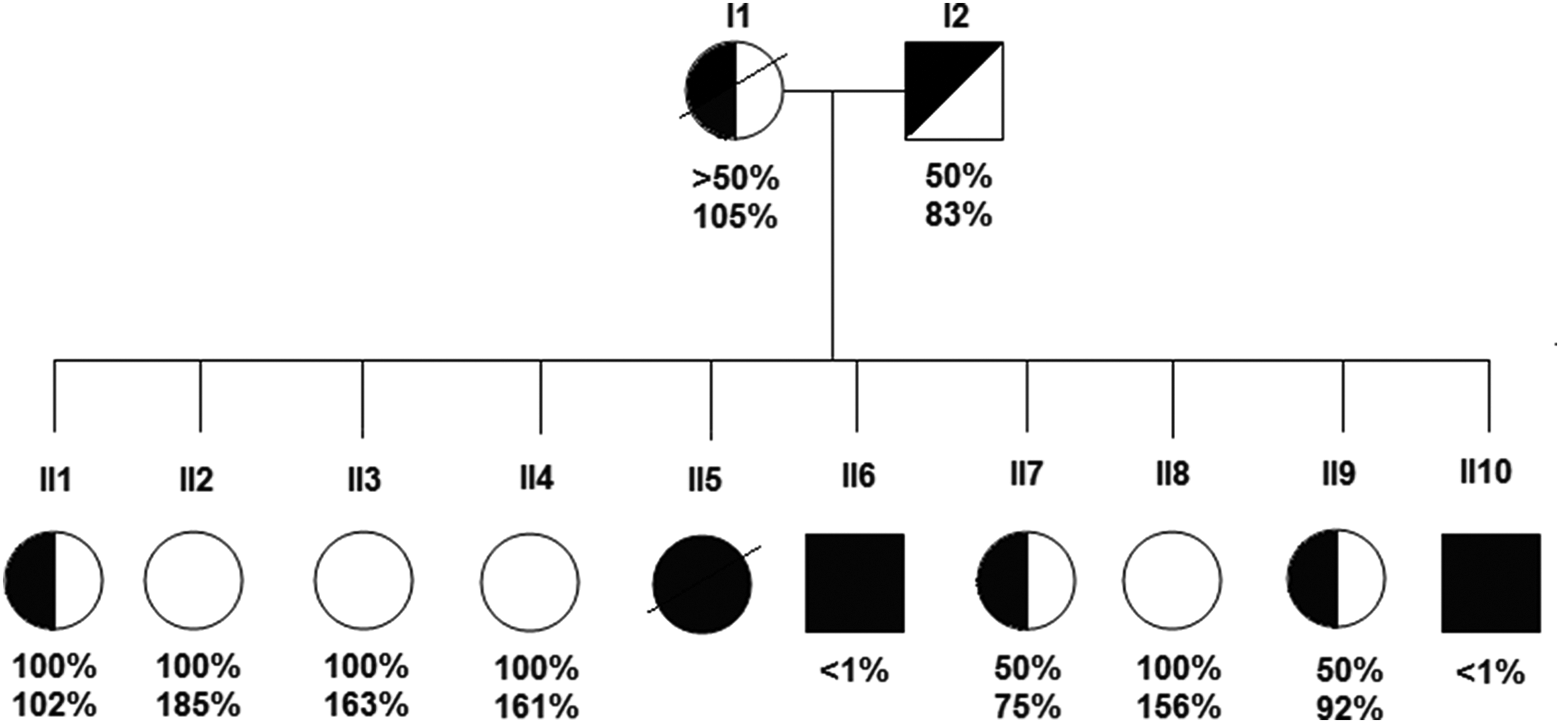
Pedigree of the family. Squares and circles indicate males and females, respectively. Upper figures refer to a disintegrin and metalloproteinase with thrombospondin type I motif, member 13 protein (ADAMTS13) activity as assessed by the immune assay (normal range 50%-200%) and lower figures are the results of the collagen-binding assay (normal > 150%).
Later, plasma levels of ADAMTS13 activity were measured by a protease assay, which consisted of 2 principal steps, first proteolysis of VWF substrate by patient’s plasma ADAMTS13 followed by quantification of digestion products or residual VWF activity.
An assay using a 1:1 (vol:vol) mixture with normal plasma detected no inhibitory antibody against the protease.
The assays were repeated in 2009 for family members II6 and II10 and again showed less than 1% activity for ADAMTS13.
Sequence analysis of DNA amplified by polymerase chain reaction showed that both family members II6 and II10 have double heterozygote mutations in exon 7 in the metalloprotease domain and exon 26 in the CUB1 (C1r/1s, Uegf, and BMP1) domain of the ADAMTS13 gene. The exon 7 frameshift mutation that leads to deletion of a single nucleotide c.717C has never been described before. The second exon 26 mutation is a c.3655C>T missense mutation leading to p.R1219W.
Hematological and biochemical data collected from patients in relation to the time interval from the last plasma infusion was analyzed using the nonparametric locally weighted scatterplot smoothing (LOESS) providing a graphical diagnostic of trend.
Family Description
The family is Arab living in the north of Israel. The parents were not consanguineous. Family members are listed in Figure 1. Family members II5, II6, and II10 were initially described in 1988. 11
Family Member I1
Family member I1 is the mother of the family. She was an obligate carrier with low levels of ADAMTS13 but had no recorded episode of TTP. She died at the age of 63 with multiple myeloma and cardiac amyloidosis.
Family Member I2
Family member I2 is the father of the family. He is an obligate carrier with low levels of ADAMTS13 but has had no recorded episode of TTP. He is presently aged 77 and has had no evidence of major cardiovascular disease apart from mild mitral valve prolapse.
Family Member II1
Family member II1 is a suspected carrier with low levels of ADAMTS13 on the collagen assay. She is now aged 53 and has had no recorded episodes of TTP. She has had no thrombocytopenia or pregnancy-related complications throughout 4 pregnancies. One further pregnancy resulted in a miscarriage.
Family members II2, II3, II4, and II8 have normal levels of ADAMTS13 and no reported relevant disease.
Family member II5 died of TTP at the age of 22 months in 1968, which was confirmed on a postmortem examination. At the age of 1, she had severe thrombocytopenia and microangiopathic hemolytic anemia associated with vomiting and diarrhea. Her condition improved after a blood transfusion but relapsed after 5 weeks when she was again transfused with blood. The fatal episode occurred some 5 months later after she went into a coma.
Family Member II6
The initial attack of TTP with severe anemia and thrombocytopenia occurred in this patient in 1968 when he was 2.5 months old. He was treated with red cell transfusions, and his condition gradually improved over a 2-month hospital stay. Two mild self-limiting attacks occurred at 4 and 13 years and then a major attack after an appendectomy at the age of 15 years. 12 He was treated again with red cell transfusions and for the first time with an infusion of FFP following which he made a prompt recovery. After this he had documented attacks of TTP almost on an annual basis which were treated by FFP. The frequency of attacks increased at the age 31 to 32 years, and he was then started on a regime of regular plasma infusions. Initially he was seen every 2 weeks, and the decision to give an infusion was dependent on the platelet count. It was later decided to give regular infusions of 1 or 2 units of plasma for every 2 weeks not dependent on this count or other biochemical testing.
In spite of this, he continues to have attacks of TTP. These may occur spontaneously after episodes of infection such as gastroenteritis. Some of these are evident in hematological and biochemical parameters taken before receiving a regular plasma infusion and are quickly aborted by the infusion without the need for hospitalization. Many are asymptomatic; others are accompanied by a variety of symptoms such as abdominal pain, dyspnoea, headache, muscle weakness, transient paresthesiae, or general lassitude, which quickly resolve after the plasma infusion.
He has also had more severe episodes requiring hospitalization. Several have occurred with a transient left hemiparesis. Computed tomography scans show evidence of small old infarcts in the basal ganglia and periventricular areas. There have also been 2 episodes of atrial fibrillation reverting to sinus rhythm with drugs and without evidence of a myocardial infarction. Echocardiography was normal. A thrombophilia screen (Table 1) shows the presence of heterozygous factor V G1291A (Leiden; FVL) and homozygous methylenetetrahydrofolate reductase (MTHFR) 677C > A. Homocysteine levels were normal, but folic acid supplements have been given.
Family Members in Relation to Thrombophilia Determinants.a
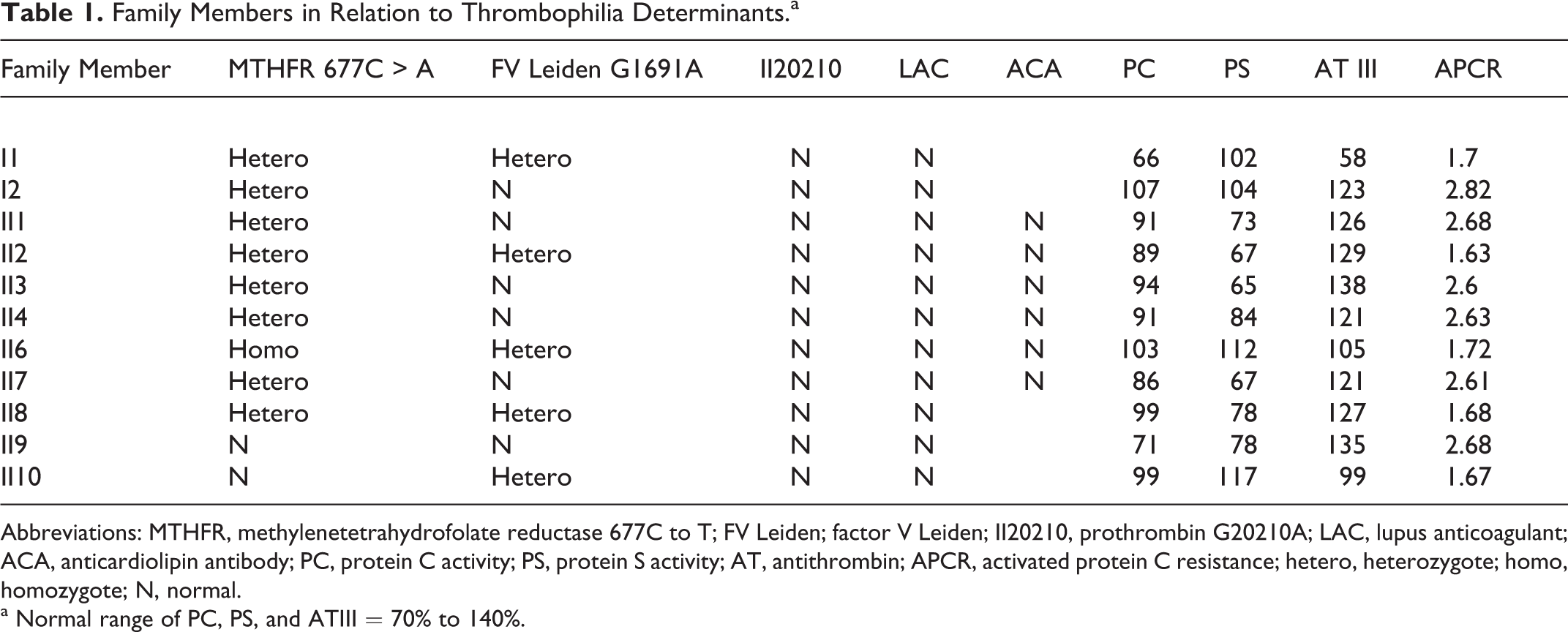
Abbreviations: MTHFR, methylenetetrahydrofolate reductase 677C to T; FV Leiden; factor V Leiden; II20210, prothrombin G20210A; LAC, lupus anticoagulant; ACA, anticardiolipin antibody; PC, protein C activity; PS, protein S activity; AT, antithrombin; APCR, activated protein C resistance; hetero, heterozygote; homo, homozygote; N, normal.
a Normal range of PC, PS, and ATIII = 70% to 140%.
From the age of 32, creatinine has been consistently elevated in the region of 1.5 mg/dL (Figure 2). Ultrasound shows a single kidney with agenesis on the right side. In 3 major attacks between the ages of 40 to 43, there were particularly high elevations of the serum creatinine reaching a maximum of 9.03 on 1 occasion. In 1 attack at the age of 40, he had an episode of renal colic with evidence of a calcified stone in the ureter. The stone later passed spontaneously.
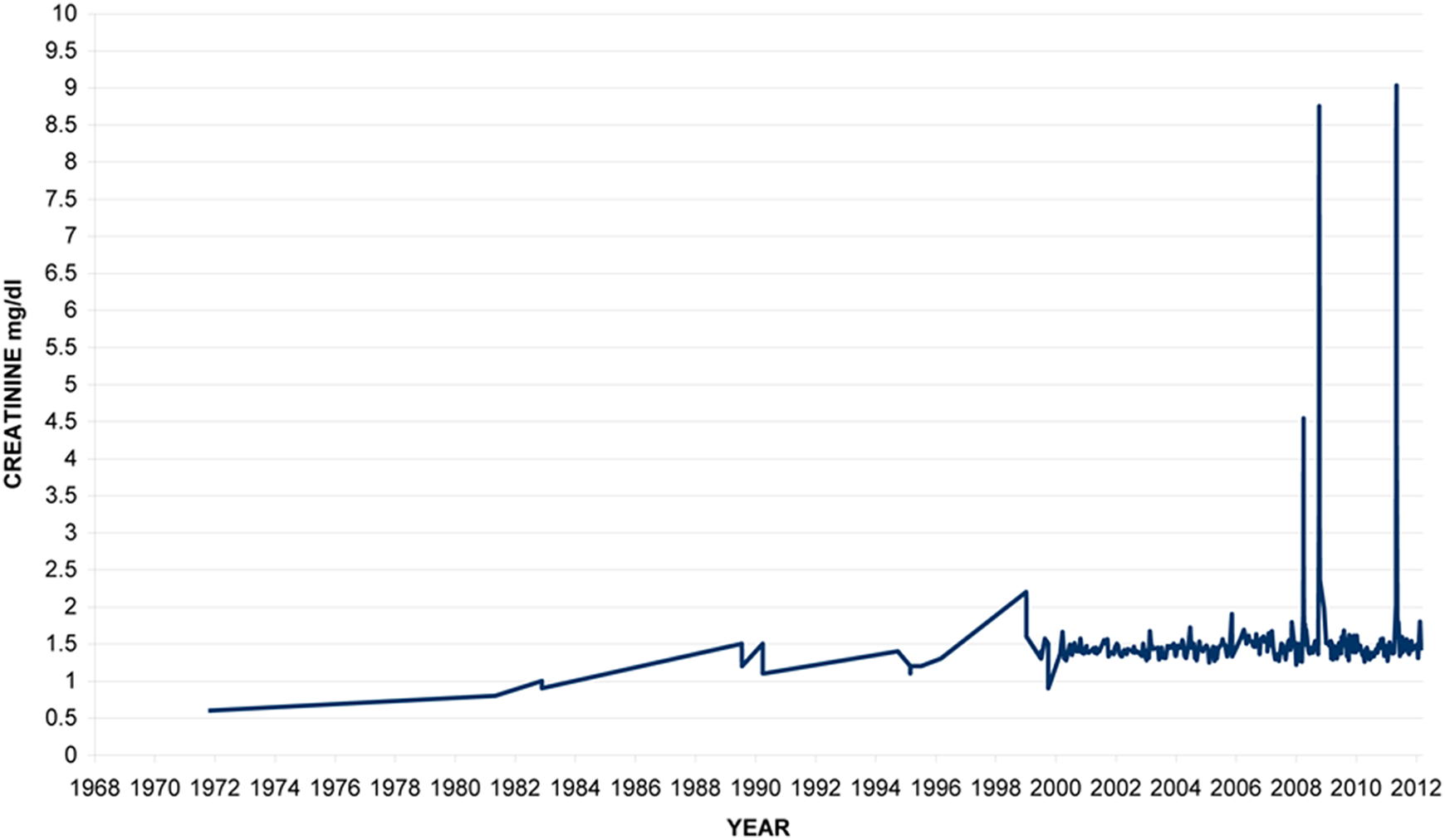
Family member II6: creatinine levels over time.
He has had at least 3 episodes of severe reactions to plasma infusions, 2 manifesting in anaphylactic shock and 1 of these requiring admission to the intensive care unit. Antihistamines are now taken before plasma infusions.
The last major attack requiring hospital admission was at the age of 43. This started in the evening 12 days after a plasma infusion with dizziness, disturbance in vision, vomiting. and a transient hemiparesis and loss of consciousness. In the initial first few hours, the platelet count fell from 71 to 24 × 109/L and then rose to 54 after 2 days and then to 158 after 6 days. Lactate dehydrogenase (LDH) levels fell quickly after the initiation of therapy, but creatinine levels continued to rise reaching a maximum of 9.03 mg/dL 3 days after the start of the attack falling to basal levels 16 days later. Following the initiation of plasma therapy, he noticed an improvement in well-being before objective changes in blood parameters. This has often been apparent in previous severe episodes, sometimes leading to a request for a discharge from hospitalization before the consent of the treating physician.
In an attempt to try and improve this patient’s management, an analysis has been made on laboratory data collected over the last 12 years taken before plasma infusion in relation to the time interval from the last plasma infusion. The results are shown in Figure 3A to C. This clearly shows that a delay beyond 13 to 15 days between plasma infusions is deleterious in relation to rises in bilirubin and LDH levels. Platelet counts fall continuously between infusions and below 150 × 109/L after day 14.
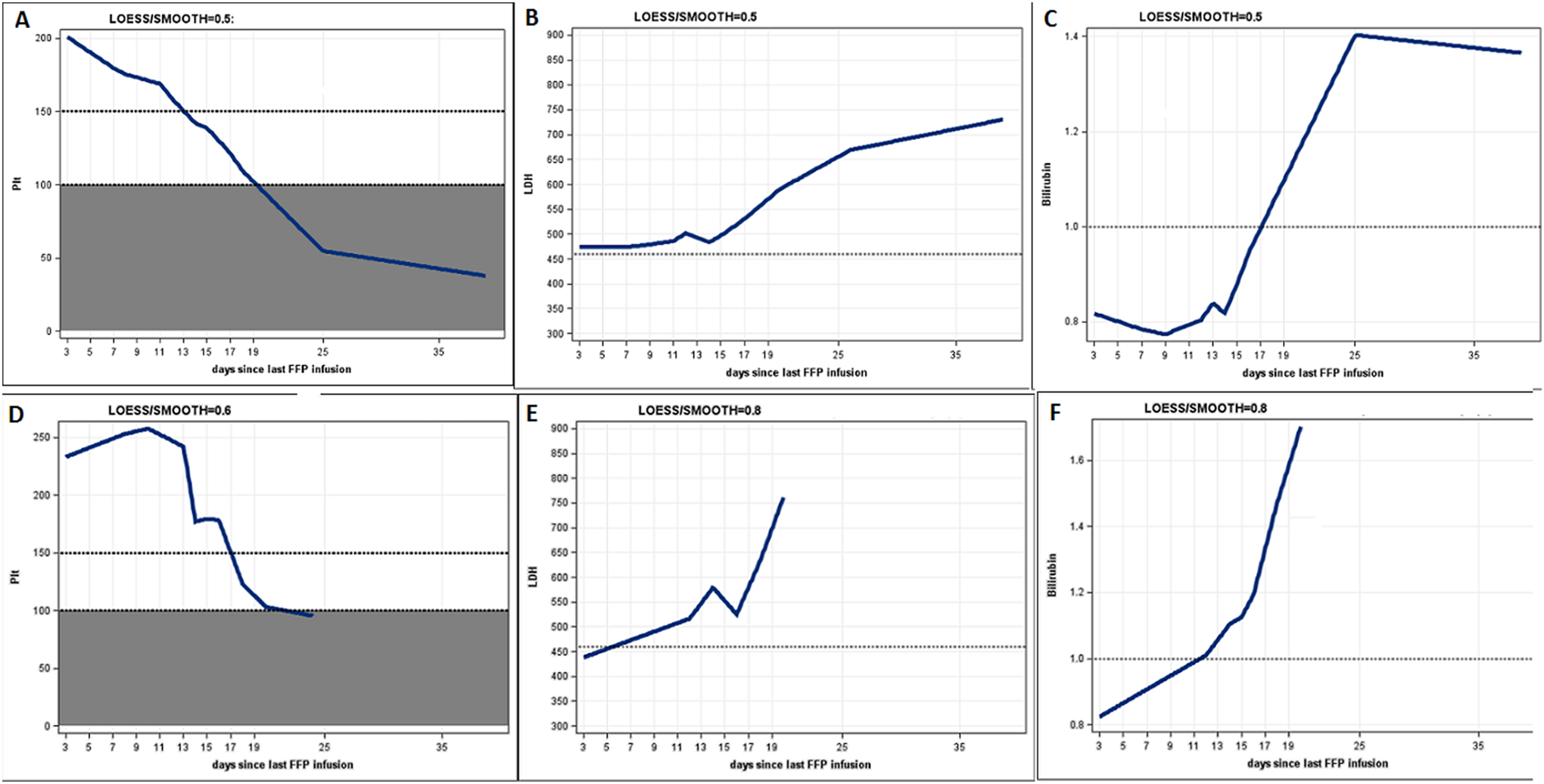
Locally weighted scatterplot smoothing (LOESS) regression analysis showing relation between platelet count, LDH and bilirubin levels, and time since last plasma infusion. The volumes of plasma infused were mainly between 1 and 2 units. Family member II6: (A-C). Family member II10: (C-E). Plt indicates platelets × 109/L. LDH, lactate dehydrogenase, U/L; bilirubin, mg/dL.
At present, this family member is aged 44 years and is receiving treatment for hypertension. He is a lawyer but has short-term memory loss and is not able to work in his profession. He has 3 children who have no medical history but have not been tested for carrier status. Two units of plasma are currently infused to him on a weekly basis.
Family Member II7
Family member II7 is a carrier with low levels of ADAMTS. She is now aged 40 and has had no recorded episodes of TTP. She has had no recorded thrombocytopenia or pregnancy-related complications such as preeclampsia or premature delivery throughout 4 pregnancies. One further pregnancy resulted in a miscarriage.
Family Member II9
Family member II9 is a carrier with low levels of ADAMTS13. She is now aged 35 years. At the age of 18 years, she was found to have a mild thrombocytopenia of 76 × 109/L with abdominal pain and iron deficiency (Hb 9.4 g/dL) but without frank evidence of TTP. In the view of the family history in her brothers, she received plasma infusions and the platelet count rose.
Further similar episodes have recurred on 2 occasions at the ages of 20 and 22 years. The last was accompanied by a raise in LDH to 815 U/L (normal range 230-460). She has had 3 pregnancies with no reported thrombocytopenia during pregnancies.
Family Member II10
This patient had his first attack at the age of 18 months, which was diagnosed as hemolytic uremic syndrome. He received a blood transfusion, and as a result his platelet count and general condition improved. A second attack occurred at the age of 3 years which was treated by blood transfusion and a third at the age of 4 years when he was given plasma and platelet transfusions. He then had more attacks of documented TTP occurring at a greater frequency of up to 11 per year. Initially, these were treated with red cell and occasional platelet transfusions. In 2 episodes, the antifibrinolytic agent hexokapron was given, and the patient went into remission. From the age of 15 years, attacks were treated with plasma infusions to which he rapidly responded. In severe attacks, the platelet count usually doubled within 1 day from less than 50 × 109/L and reached up to 9 times the pretreatment value by the fifth day. Initially, whole plasma was given and later cryo-removed plasma.
At the age of 18 years he underwent an elective splenectomy in order to try and reduce the increasing frequency of attacks. The operation was performed without complication with prophylactic plasma infusion. This procedure was not, however, efficacious, and he continued to have episodes of TTP. In a 17-month postoperative period, he had 8 major and 7 minor attacks. He was found to have had a pulmonary embolus and was treated with enoxaparin. After 6 months, this therapy was stopped, and low-dose warfarin was substituted. A short time afterward, evidence of fresh emboli was found, and it was decided to continue oral anticoagulant therapy with warfarin in therapeutic doses for life. A thrombophilia profile showed that he was heterozygous for FVL (Table 1).
For the next 5 years with oral anticoagulant therapy, there were less frequent episodes of TTP (1-4 per year). Attacks were, however, of a severe nature, several with marked neurological features. As can be seen in Figure 4, platelet counts were higher postsplenectomy. There were higher rebound counts after treatment, and attacks were apparent with a higher platelet count. Hemoglobin levels have also been higher postsplenectomy usually between 16 and 18 g/dL. There was no evidence of pulmonary hypertension.
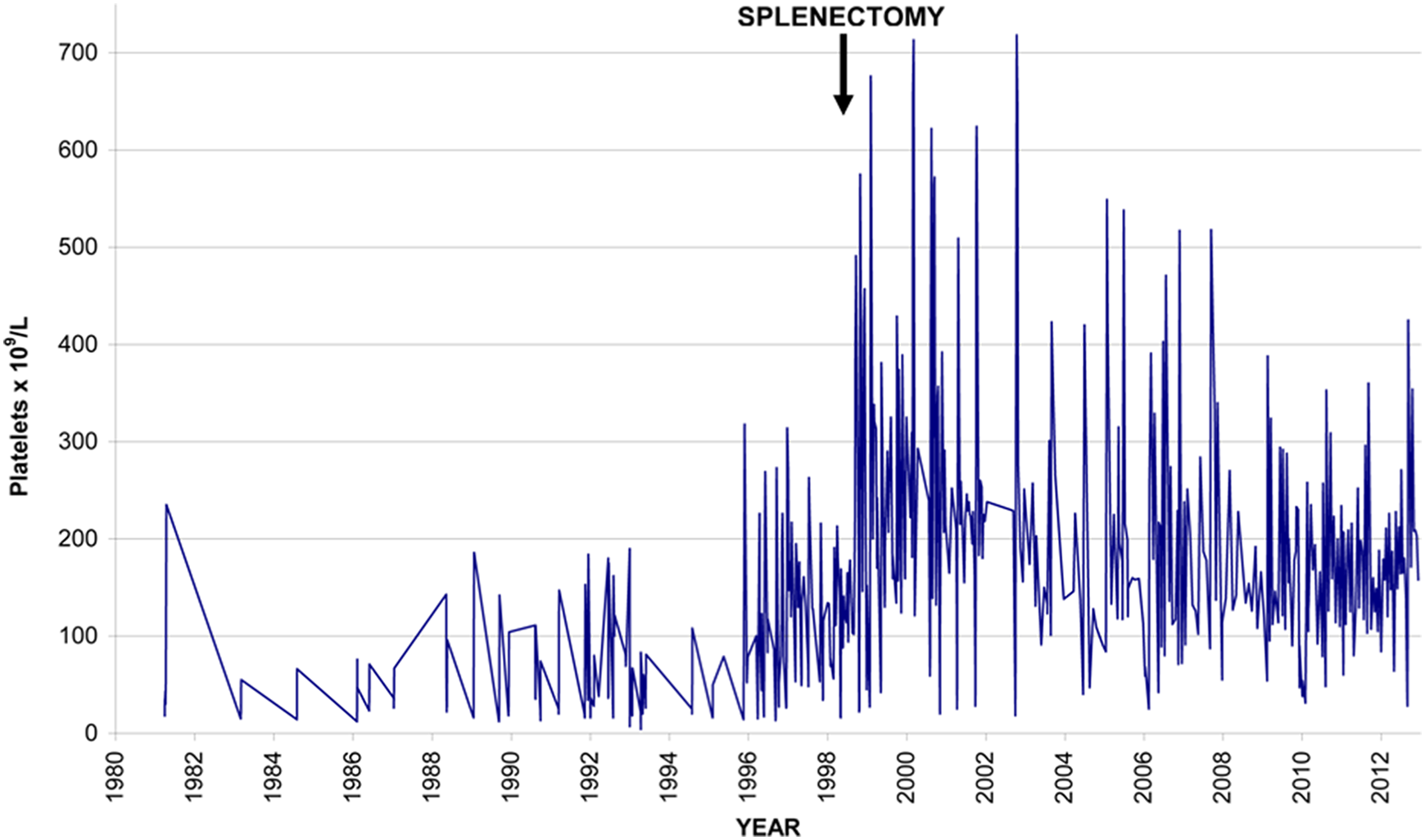
Family member II10: platelet counts.
In order to try and reduce the incidence of severe attacks, prophylactic plasma infusions were suggested as in his elder brother. At first he was reluctant, but in the last 2 years he agreed to receive these. During this period, he had only 1 major attack requiring hospital admission, but in many instances, there have been reductions in the platelet count and elevations in LDH or bilirubin indicative of a minor or impending major attack.
The last major attack was at the age of 32 with acute renal failure. Figure 5 illustrates the response to plasma infusions given daily for the first 5 days of this and then on days 21 and 36. Initially, here there was a fall in platelets and rise in LDH for several hours despite initiation of treatment. The creatinine continued to rise for several days despite improvement in LDH and platelets and did not return to normal levels for 7 weeks.
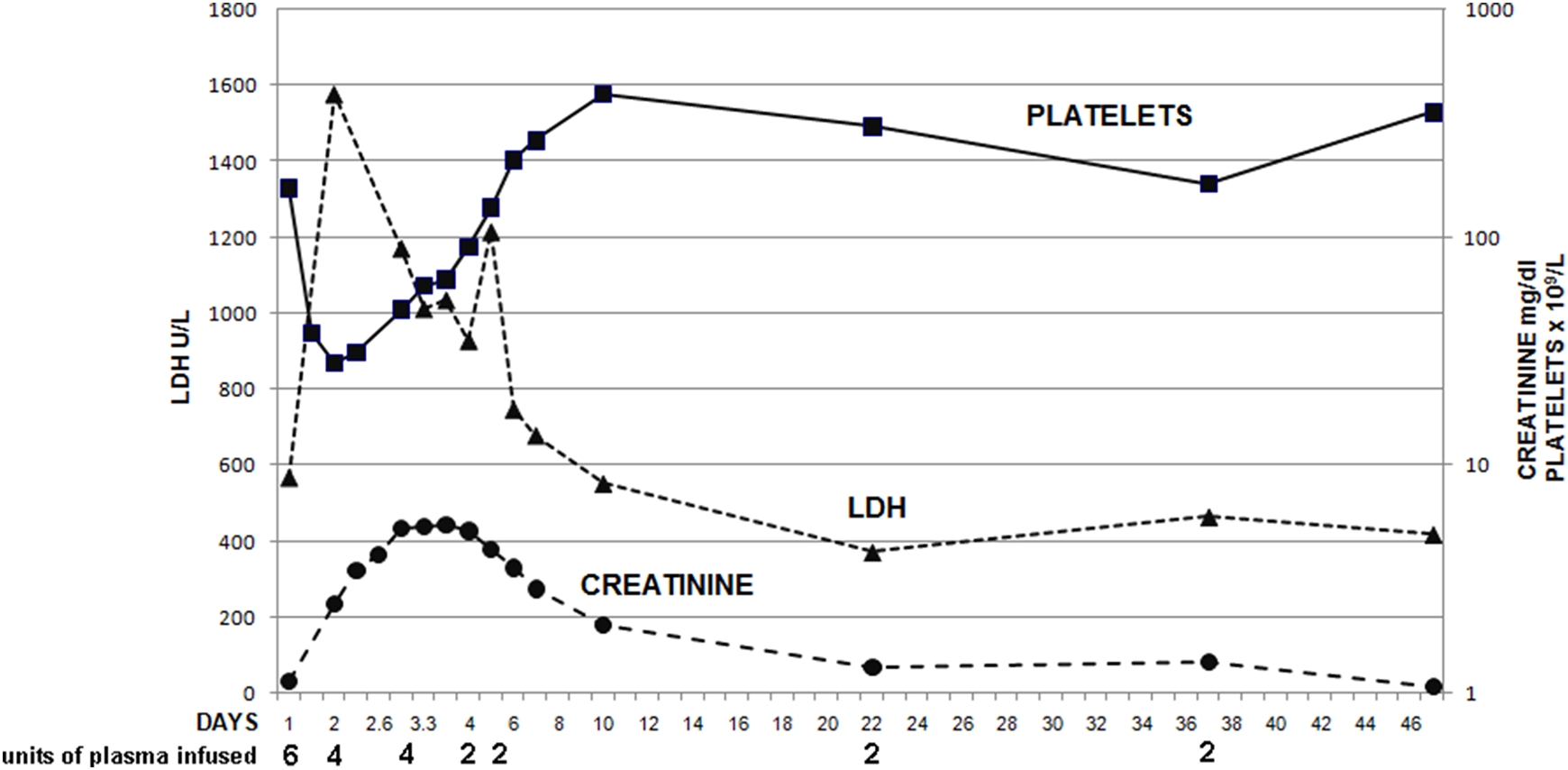
Family member II10: response to treatment in a major attack.
Analysis of his parameters over the last 2 years according to the nonparametric LOESS smoother is shown in Figure 3D to F. Platelet counts started to fall at 13 days but from a higher level than in his brother. The LDH and bilirubin started to rise earlier at 7 days.
Creatinine levels were not included in this analysis as they were often normal at the outset of a major attack and rose later.
At present, this family member is aged 33 years.
Discussion
The child (II5) died in 1968 before the modern era in the treatment of TTP. The diagnosis was, however, confirmed by a postmortem examination. This diagnosis influenced the treatment of the later siblings who developed features of the disease.
Both the compound heterozygous cases (II6 and II10) had severe attacks early in life at the age of 2.5 months and 18 months, respectively. Treatment at that time was primitive and did not include plasma infusions. They were fortunate to survive these episodes, and it is possible that plasma in red cell and platelet transfusions helped to supply ADAMTS13. Platelet transfusion and antifibrinolytic agents are today theoretically contraindicated in TTP, but they had no recognizable adverse effects on these cases. The evidence for harm from platelet transfusion is uncertain. 13
They correspond to an early-onset phenotype, 7 but then went through long periods without attacks. Attacks started to become more frequent in adolescence, sometimes associated with infection or trauma or sometimes without any apparent cause. As these patients become older and the level of VWF rises, one might expect more frequent attacks. 14 As they become older, atherosclerosis may become more apparent as has been seen in ADAMTS13-deficient mice. 15 There has been no clinical evidence of accelerated atherosclerosis in the 2 brothers.
Although splenectomy may be beneficial in acquired, resistant, or relapsing TTP 16,17 and has been reported to be successful in a case of USS, 10 it was not so in II10. The thromboembolic complications were probably related to the continued episodes of intravascular hemolysis in a postsplenectomy state, and this has necessitated lifelong oral anticoagulant therapy. Warfarin therapy did not affect the frequency of TTP attacks in this patient but has been administered without further thromboembolic events and with no side effects.
The presence of the FVL mutation may also contribute to a hypercoagulable state in both II6 and II10. This association has not been reported before in USS. FVL has been suggested to be related to acquired TTP with normal ADAMTS13 levels, 18 although this has been queried. 19
Atrial fibrillation in II6 may be related to myocardial damage that has been reported in acquired TTP. 20 –24 Oral anticoagulant therapy is being considered for this patient in the view of this together with the presence of the FVL mutation and evidence of cerebral infarction.
In patients with USS needing prophylactic infusions of plasma, it has been suggested that an infusion frequency of every 2 to 3 weeks would be sufficient to prevent attacks. 25 Although the half-life of ADAMTS13 is only 2 to 3 days, 26 low levels may be sufficient in preventing an attack. In patients II6 and II10, we analyzed the relation between parameters with a LOESS regression model analysis indicating an attack and periods between plasma infusions. By 14 days, generally, the platelet count was already falling and the LDH rising, which indicates the need for a more frequent transfusion regime.
In managing these patients, one is naturally concerned that although the attacks are quickly reversible with plasma infusion, permanent disabilities ensue. This is apparent in II6 who has irreversible renal and cerebral damage. The former may be exacerbated because of a unilateral kidney or may be part of the natural history of the disease as was seen in Schulmans original patient. 27
Finally, the deep psychological problems that affect II6 and II10 should not be underestimated. Although no major depression or cognitive deficits have been documented, they have to live daily with the constant knowledge that they could have a potentially fatal attack of TTP. The use of recombinant ADAMTS13 and the potential for self-administration and home administration as in Hemophilia are eagerly awaited.
Footnotes
Acknowledgments
The authors would like to thank M. Furlan and J.A. Kremer Hovinga in The University of Bern, Switzerland for their support, for measuring ADAMTS13 protein levels and for molecular analyses. They would also like to thank Naama Schwartz for statistical analyses.Authors’ Note
MB initially diagnosed and managed the family initially. All authors were concerned with their management in later years. MB and YC wrote the article and collected data. IG, AA, GS, and DC-M analyzed the data and reviewed the manuscript.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.