
Abstract
Acquired hemophilia A (AHA) is a rare bleeding disorder caused by the autoantibody directed against factor VIII in patients without previous history of a bleeding disorder. We retrospectively analyzed the characteristics and outcomes of 49 patients with AHA diagnosed in our center from February 1994 to October 2012. Twenty-four patients with acute bleeding episodes were treated with prothrombin complex concentrate (PCC) at a relative low dose of 30 to -50 U/kg/d and achieved good outcomes without any adverse reaction. Corticosteroids alone or in combination with cyclophosphamide were used as the first-line therapy to eradicate the inhibitors. In 39 evaluable patients, 35 (89.7%) achieved complete remission (CR). This study demonstrates that when bypassing agents such as recombinant activated factor VII and activated PCCs are not affordable or available, low dose PCC is effective and safe to control acute bleeding in patients with AHA. First-line therapy achieved good outcomes with a CR rate of 89.7%.
Introduction
Acquired hemophilia A (AHA) is a rare bleeding disease resulting from the production of autoantibodies directed against coagulation factor VIII (FVIII) in nonhemophilic patients. It often occurs in elderly persons with an incidence of about 1.5 per million populations per year. 1 Hemorrhage can be life threatening and often occurs spontaneously or after trivial trauma or surgery. The most frequent symptom was hemorrhage of skin, muscles, or soft tissues. Hemarthrosis are uncommon in AHA that differs from congenital hemophilia. 1,2 Severe bleeding occurs in a majority of patients with a high rate of mortality ranging from 9.7% to 33%. 2,3,4 Early recognition, rapid diagnosis, and proper treatment are very important to improve the outcomes of bleeding, but many clinicians in China lack awareness of this disease. To improve our knowledge of this disease, we retrospectively analyzed 49 patients with AHA referred to our hospital.
Patients and Methods
Patients
From February 1994 to October 2012, 49 patients with AHA were diagnosed and treated in our hospital. The detailed data on clinical history, underlying conditions, and physical examination were recorded. No patients had congenital clotting factor deficiency or a family history of bleeding disorders. All patients were diagnosed based on the acute or recent onset bleeding and a prolonged activated partial thromboplastin time (APTT) and a decreased activity of FVIII, at the time of presentation to the clinic. Factor VIII inhibitor was demonstrated with the Bethesda assay. 2
Laboratory Assays
Complete blood counts, liver and renal functions, test for hepatitis B virus , hepatitis C virus, and HIV, levels of immunoglobulin, thyroid function test, rheumatoid factor, antinuclear antibody, anti–double-strand DNA, tumor markers, chest radiograph, and B ultrasonic wave were performed in all patients. Screening coagulation tests including prothrombin time (PT), APTT, antithrombin III activity, fibrinogen, and
Treatment
Treatment of Acute Hemorrhages
Prothrombin complex concentrate (PCC; Prothoraas, Shanghai RAAS Blood products Co LTD, China; or Kangshuning, Hualan Biological Engineering, Inc, China) was used for acute bleeding episodes at a dose of 30 to 50 U/kg per d. Other hemostatic therapy consisted of FVIII concentrates, fresh frozen plasma (FFP), and topical hemostatic therapy. Packed red blood cells (RBCs) were used in patients with severe anemia.
Inhibitor Eradication
All patients received immunosuppressive therapy to eradicate the inhibitors of FVIII. Corticosteroid was given at a dose of prednisone 0.5 to 1 mg/kg per d tapered gradually based on response or adverse effect. Cyclophosphamide was given at a dose of 100 mg every day (qd) or 200 mg every other day (qod) intravenously (iv) and discontinued when the white blood cell (WBC) counts were below 4 × 109/L. Azathioprine was used as the maintained therapy at 50 to 150 mg/d orally when corticosteroid was tapered and cyclophosphamide was discontinued.
Second-Line Therapy
A single dose of rituximab of 375 mg/m2 or a dose of 100 mg/m2/week given for 4 weeks was used when the first-line immunosuppressive therapy failed.
Treatment Response
Complete remission (CR) was defined as the disappearance of hemorrhagic symptom accompanied by a normal FVIII level (≥50%) and undetectable FVIII inhibitor. 1 If CR was not achieved, FVIII:C increased to ≥20%, and the decrease in inhibitor titer by >50% was considered as partial response (PR). The response to PCC and other hemostatic therapy was evaluated by clinical manifestation including the arrest of hemorrhage (such as hematoma absorbed, the ache relieved, the ecchymosis and hematuria disappeared), without new bleeding episode.
Follow-Up
All patients were followed up till January 2013.
Results
Patient Characteristics
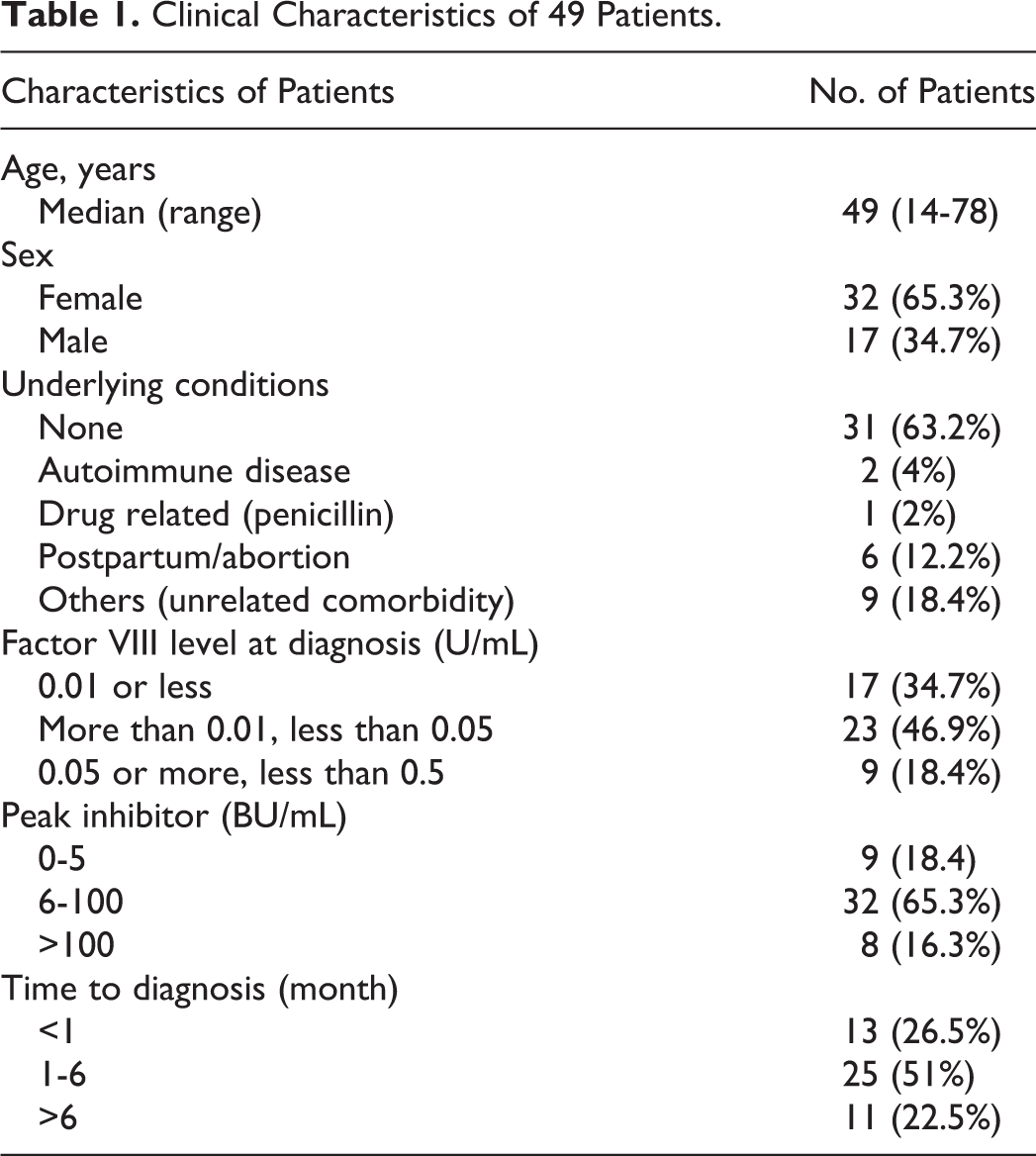
The clinical characteristics of 49 patients (men 17 and women 32) are shown in Table 1. The age and sex distribution are shown in Figure 1. Clinical symptoms are shown in Figure 2. Bleeding occurred spontaneously in a majority (46 of 49) of patients. In 3 patients, bleeding was secondary to surgery or trauma. The median interval from bleeding onset to diagnosis was 4 months, the longest interval being 3 years.
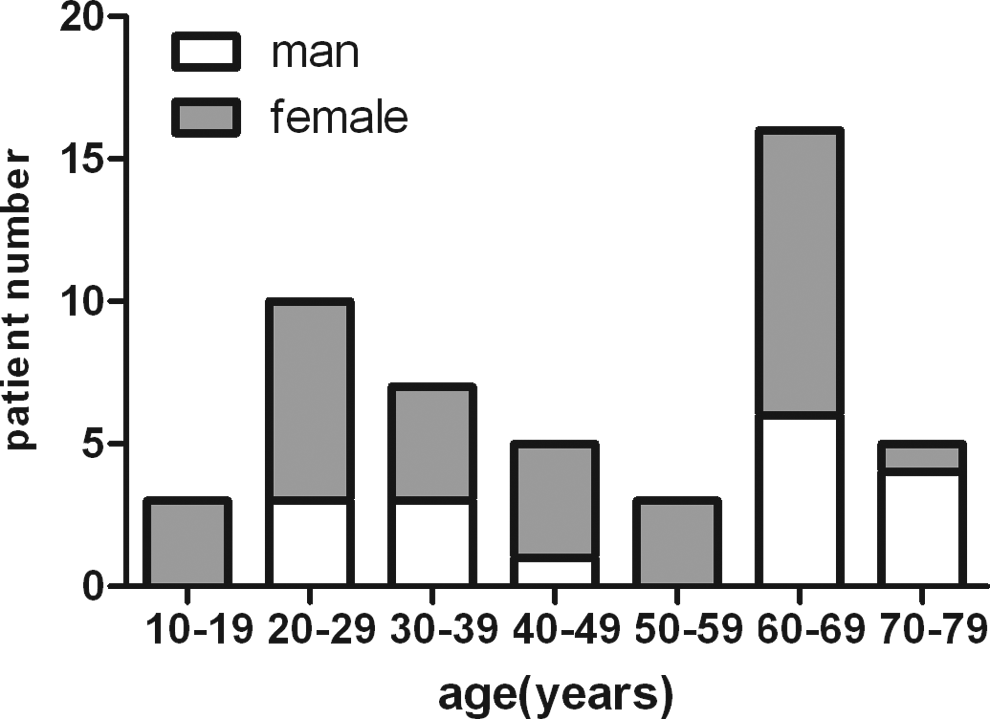
Age and sex distribution.
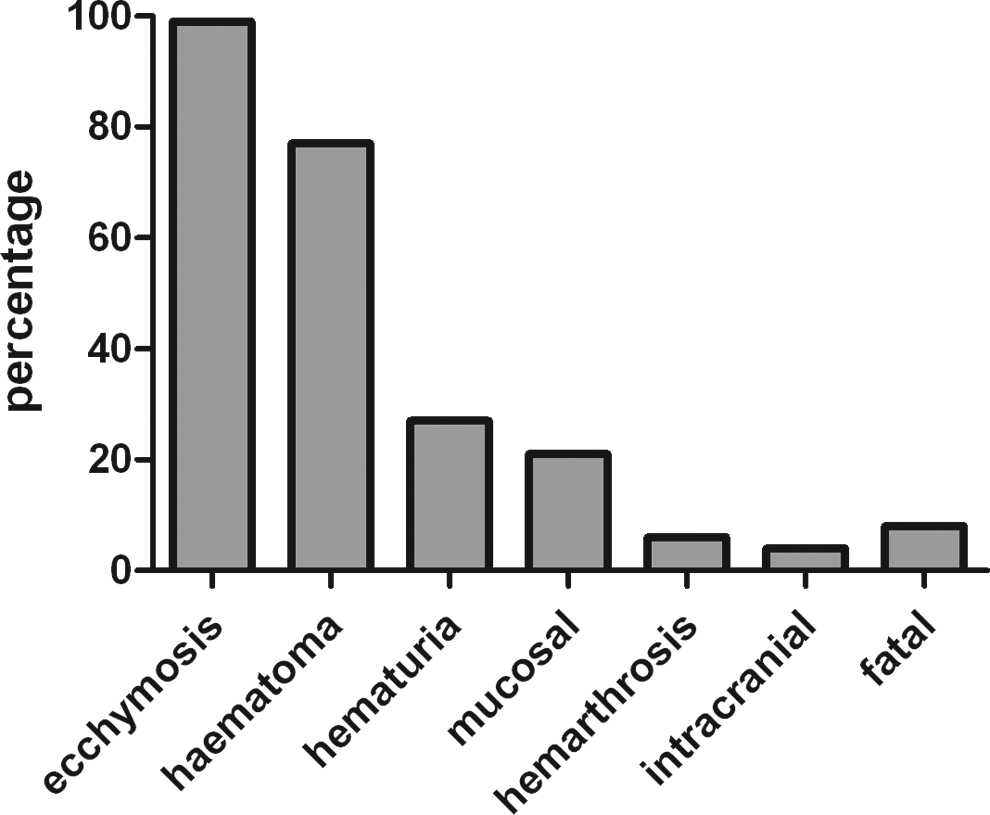
The percentage of bleeding episodes in patients. Most patients have more than 1 type of bleeding.
Clinical Characteristics of 49 Patients.
Etiology or Comorbidities
Most patients (31 of 49, 63.3%) had no associated disease or underlying medical condition. In 6 patients, the inhibitor occurred during the postpartum period (n = 4) or following an abortion (n = 2). Among them, 1 patient had ecchymosis during pregnancy, had severe postpartum hemorrhage, and had undergone total abdominal hysterectomy to control hemorrhage 3 years before her presentation at our hospital. Unfortunately, she lost to follow-up finally. Inhibitors in 2 patients were associated with autoimmune disorders. One of patients had been diagnosed with idiopathic thrombocytopenic purpura and was treated before 9 months. He presented with severe bleeding symptom but had normal platelet counts when referred to our hospital. The other had myasthenia gravis and was treated with thymus resections prior to 8 years, followed by low-dose corticosteroid until AHA was diagnosed. One patient presented with ecchymosis during the course of penicillin injection. Nine patients had comorbidities including coronary artery disease, chronic hepatitis B, chronic cholecystitis, hypertension, and type 2 diabetes.
Laboratory Findings
Laboratory results are summarized in Table 1. All the 49 patients had normal PT and normal or elevated platelet counts. Mean APTT was 99 seconds (range 61.2-180 seconds). All patients had decreased FVIII:C level and normal levels of other factors (data not shown). All patients were positive in inhibitor screening test. Forty patients had high-titre inhibitor (>5 BU/mL). 7 The median peak titer of the inhibitor was 22 BU/mL (range 1.5-825). vWF-Ag range from 0.68 to 7.93 U/mL (reference = 0.5-2.0 U/mL; data not shown). All patients were negative for lupus anticoagulant.
Treatment and Outcomes
The treatment and outcomes were shown in Table 2. Because recombinant activated FVII and activated PCC were not available or affordable, we administered PCC at a dose 30 to 50 U/kg per d to stop acute bleeding episodes. Twenty-four patients with acute hemorrhages were treated with PCC, ranging from 1 to 19 days depending on the severity of bleeding. The PCC was effective in 23 patients and showed no adverse events. Case 12 received FVIII concentrates and PCC for 4 days but showed less response and presented with subcutaneous hematoma and epistaxis intermittently, and she could not afford further hemostatic therapy. Case 7 responded well to the treatment in that the hematoma was absorbed and the ecchymosis disappeared, but suddenly she died of intracranial bleeding at week 7. Case 21 was subjected to severe hematuria and received FVIII as initial hemostatic therapy, but the hematuria was not alleviated until PCC was used for 7 days.
Laboratory Results, Treatment, and Outcomes of 39 Evaluable Patients.
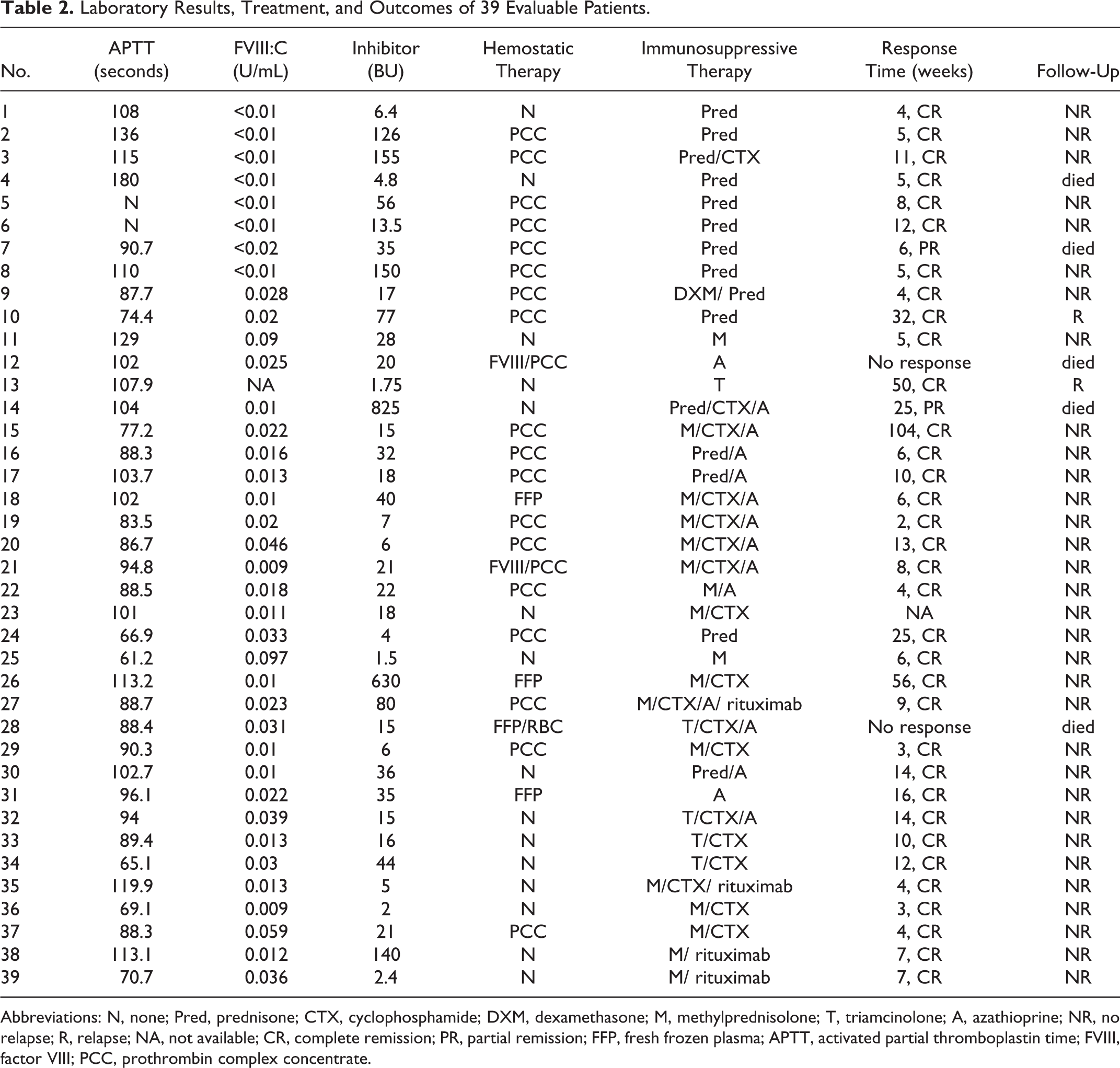
Abbreviations: N, none; Pred, prednisone; CTX, cyclophosphamide; DXM, dexamethasone; M, methylprednisolone; T, triamcinolone; A, azathioprine; NR, no relapse; R, relapse; NA, not available; CR, complete remission; PR, partial remission; FFP, fresh frozen plasma; APTT, activated partial thromboplastin time; FVIII, factor VIII; PCC, prothrombin complex concentrate.
To eradicate the inhibitors, all patients received immunosuppressive therapy when the diagnosis was confirmed. All patients were followed up till January 2013 and the median time was 57 months (range 12-216 months). As shown in Table 2, 37 of 39 evaluable patients (6 patients were lost to follow-up, 4 patients are currently in treatment) were treated with corticosteroid alone or combined with cyclophosphamide and/or azathioprine. Case 27 was a 29-year-old woman with myasthenia gravis, her inhibitor titer increased even though corticosteroid combined with cyclophosphamide were administered for 1 month. Thus, rituximab was given as a second-line therapy. Before referring to our hospital, case 35 presented with persistent hematuria, although he was given corticosteroid, cryoprecipitate, and FVIII. Thus, rituximab (375 mg/m2) was given once. These 2 patients maintained the first-line therapy after treatment of rituximab and achieved CR in 1 and 2 months, respectively. In 39 evaluable patients, 35 (89.7%) achieved CR. The median time to CR was 8 weeks (range = 2-104 weeks). The response time was not related to FVIII:C and inhibitor titer. Two patients (case 10 and 13) had a relapse after 4.5 months and 1 year, respectively; but they achieved second CR after the second cycle of therapy. In our study, 5 patients died. In particular, case 7 responded well to the therapy with disappearance of hemorrhage and achieved PR in 6 weeks but died of intracranial hemorrhage at week 7. Case 12 was a 72-year-old woman with type 2 diabetes who was given azathioprine alone. After 1 year of treatment, she did not achieve CR and presented with pancytopenia and died of sepsis and multiple organ hemorrhage. Case 28 died of infection and multiple organ hemorrhage. Case 14 refused to standard treatment before CR and died of intracranial hemorrhage. One patient (case 4) died of lung cancer without AHA relapse 2 years after treatment but no evidence for lung cancer was found at the diagnosis of AHA.
Discussion
This study described the characteristics and treatment regimens of AHA based on the largest series in China. Acquired hemophilia A is a rare bleeding disorder that occurs predominantly in adults. The incidence increases with age, and in some studies the median age was 78 years. 1,8 The age distribution is biphasic, with a major peak in patients of age 68 to 80 years and a second frequency peak occurring between 20 and 30 years, due to postpartum-related inhibitors. 9,10 In our cohort, the second frequency peak was consistent with previous studies but the median age (49 years) and the major peak age were much younger than that reported in other studies. We postulate that referral bias may have some effects on this cohort. Conceivably, in contrast to elderly patients, young people with AHA were more likely to be referred. Therefore, this report may not represent the whole clinical spectrum of AHA in our country.
The autoantibody against FVIII is the most common spontaneous inhibitor to coagulation factors, 11 but the etiology of the production of inhibitor remains unknown. It was reported that underlying diseases were identified in half of the patients including autoimmune diseases, postpartum period, solid tumors, infections or drug-related allergy (especially penicillin). 12 –14 However, we found much fewer cases (9 of 49) had associated diseases than other studies. It is likely that the percentage of idiopathic AHA is higher than we had presumed.
The main challenge of dealing with patients having AHA appears to be the early recognition and prompt diagnosis. Any acute or recent bleeding symptoms without a previous history of bleeding diathesis and an unexplained isolated prolonged APTT suggest the diagnosis of AHA. 15,6 Actually, most of the patients initially were referred to local hospitals where only16% of the diagnosis was confirmed. The longest time from bleeding manifestation to diagnosis was 3 years, thus exposing the patients to a high risk of severe bleeding for a long time. This situation may be due to 2 aspects. First, many doctors have little knowledge about AHA and could not recognize it when encountering a patient with an isolated prolonged APTT. Second, the FVIII:C and the inhibitor titer could not be measured in many local hospitals due to their limited facility.
The 2 main goals of treatment for AHA are to control bleeding and to eradicate the inhibitor. Although bypassing agents such as recombinant activated FVII (rFVIIa) or activated PCC (aPCC) are recommended as optimal antihemorrhagic treatments, 15,16 it is not available or not affordable for Chinese patients.
The PCCs have been used since 1970s to treat patients with inhibitors. At that time, it was recognized that the therapeutic efficacy of PCC products appeared to be related to the presence of small amounts of activated factors. 17 Therefore, aPCC was developed and became a new regimen for patients with inhibitors. 18 Then in the early 1980s, several controlled trials demonstrated the efficacy of PCCs and aPCCs in the management of acute bleeding episodes. 19 –22 Lusher et al compared the PCC with aPCC from a same manufacturer and found no difference in efficacy between products (PCC 50% vs aPCC 51.7%). 21 Consistently, our results showed that low-dose PCC is an effective and safe strategy to control acute bleeding. Although current PCC products differ from that of the old generation, some factors are activated during production unavoidably. We presume that these activated factors play a key role of hemostasis. The major drawback of PCCs is the risk of thrombotic complications. Because heparin is added into these 2 PCCs that we used and because of the relatively low dose, no thrombotic events were found. Although PCCs has been discontinued in the European countries in recent years, we consider that low-dose PCC is a reasonable choice for hemostatic treatment where rFVIIa and aPCC are not available or not affordable. It should be mentioned that repeated massive doses of PCCs should be avoided to lessen adverse events.
Case 7 was a 65-year-old woman, her FVIII:C and inhibitor were 0.02 U/mL and 35 BU/mL at diagnosis and were 0.21 U/mL and 4.5 BU/mL, respectively, after prednisone therapy for 7 weeks. Although the hematoma was absorbed and the ecchymosis disappeared, she died of intracranial hemorrhage suddenly after the last assay of FVIII:C at week 7. It was unknown whether she had some undetected cerebral vascular abnormality.
Rituximab was reported to be an effective option for the treatment of the patients with AHA refractory to standard therapies. 23 In this report, 4 patients received rituximab as the second-line therapy because the inhibitor titer continued to increase with the first-line therapy. The inhibitor titer decreased obviously in 2 weeks after using rituximab. They were then maintained on the first-line therapy and achieved CR within 2 months. However, 4 cases are insufficient to testify the efficacy of rituximab.
Delgado et al revealed by meta-analysis that there are 3 prognostic factors independently associated with the clinical outcomes of AHA: related conditions, achievement of CR, and age. 24 Indeed, the prognosis of AHA depends greatly on the side effects of long-term immunosuppression especially in elderly patients. 25 In our study, 4 patients who died of hemorrhage were older than 60 years and did not achieve CR. They had other comorbidities such as diabetes, hypertension, and coronary artery disease. Two of them had neutropenia-associated infections, which may adversely aggravate hemorrhage. Consequently, we feel that the comorbidities and the side effect of the immunosuppressive agent deserve more attention in older patients. The question whether rituximab that is more tolerable with reduced toxicity should be considered as the first-line therapy needs further studies.
Footnotes
Authors’ Note
The authors would like to thank Prof. Man-Chiu Poon (University of Calgary, Canada) for critical review of the manuscript. The authors stated that they had no interests which might be perceived as posing a conflict or bias.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported in part by grants of National Natural Science Foundation of China (81070397 and 81170474), Ministry of Science and Technology (2011ZX09302-007-04), Ministry of Health (201202017), Tianjin Municipal Science and Technology Commission (10JCZDJC19700 and 12JCQNJC08000) and Bayer hemophilia Award.