
Abstract
It is implicated that diabetic patients are more resistant to aspirin therapy than patients with other diseases or healthy individuals. We evaluated the inhibitory effects of aspirin on aggregation and the cyclooxygenase activity of platelets of 10 patients with severe type-2 diabetes mellitis (DM) and compared the results with those of healthy individuals. Although platelet aggregation had a tendency to be more resistant to aspirin with the DM group, there was no significant difference in half maximal inhibitory concentration 50 values of aspirin on the cyclooxygenase activity between the patients with DM and healthy individuals. Thus, the residual platelet aggregability uninhibited by aspirin appears to be independent of the cyclooxygenase activity. Since adenosine diphosphate (ADP) receptor blocking almost completely inhibited the residual platelet aggregability, it is suggested that hyperreactivity to ADP is more prevalent in patients with DM.
Introduction
It is widely established that antiplatelet therapy, including aspirin administration, has reduced the risk of cardiovascular events by approximately 25% (odds ratio) in a number of large-scale clinical studies. 1,2 The antiplatelet effect of aspirin is attributed to the inhibition of platelet cyclooxygenase-1 (COX-1) by acetylation of its serine 529, which results in a loss of thromboxane A2 (TxA2) production in platelets. 3 –5 However, there is an increasing body of recent evidence, which suggests insufficient effects of aspirin, referred to as “aspirin resistance.” 3,4,6 –10 The concept of aspirin resistance has been confusing due to differences in definitions and evaluation methods. 3,4 Some studies defined aspirin resistance as failure to inhibit platelet functions, such as platelet aggregation. Other studies used the production of thromboxane B2 (TxB2), a stable metabolite of TxA2, as a marker to judge aspirin resistance. Recent studies, however, have defined aspirin resistance as the failure of aspirin to suppress the occurrence of vascular events in clinical settings.
It is well known that patients having diabetes mellitus (DM) are highly susceptible to vascular events, and hence antiplatelet therapy is considered one of the key strategies to managing diabetic patients. 11,12 Despite the proven benefits of aspirin therapy in the general population, a series of reports have suggested the poor efficacy of aspirin both in primary and in secondary prevention studies for patients with type-2 DM (T2DM) with various vascular diseases. 13 –19 Several mechanisms have been proposed, which account for aspirin resistance in patients with T2DM.
One of the theories is that patients with T2DM, when compared with DM-free patients, may have more accelerated platelet turnover that results in introducing newly formed, nonaspirinated, and therefore, TxA2-producible platelets into bloodstream. 20 It is of particular interest that Winocour et al have suggested that conformational changes due to glycation or acetylation of platelet membrane proteins may cause decreased aspirin penetrance into platelets. 21 Watala et al have also suggested that increased protein glycation, possibly involving COX-1, may result in decreased aspirin-mediated acetylation of COX-1 which leads to aspirin resistance. 22 The hypothesis that protein glycation interferes with the effect of aspirin on COX-1 may explain the higher prevalence of aspirin resistance among the diabetic population. If this hypothesis proves to be true, more stringent control of the blood glucose level for diabetic patients will be required.
This study was thus planned to evaluate the validity of these glycation-related hypotheses, proposed by Winocour et al 21 and Watala et al. 22 Most of the previous studies dealing with aspirin resistance have evaluated the effects of 1 or 2 oral doses of aspirin on platelet functions, such as platelet aggregation (ex vivo assessment), or production of arachidonic acid metabolites, such as TxB2 (in vivo assessment). These studies attempted to draw a conclusion on the efficacy of aspirin on COX-1. With these experimental designs, however, it is not possible to accurately assess the degree of COX-1 inhibition and/or individual differences. It is conceivable, though, that the efficacy of aspirin in diabetic patients is only partially abrogated by glycation, and the subtle differences may not be clearly detected. Thus, it is imperative to assess the effects of varying doses of aspirin on COX-1 in vitro, and to determine the half-maximal inhibitory concentration (IC50) values of aspirin on COX-1.
Therefore, the objective is to determine whether there is any difference in the IC50 value of aspirin on COX-1 between healthy patients with individuals and severe T2DM without antiplatelet therapy.
Methods
Inclusion Criteria of Patients with T2DM and Healthy Individuals
Patients with T2DM who were not under aspirin therapy or any platelet-affecting agents were enrolled in this study. In order to elucidate the effect of COX-1 glycation, we intentionally recruited patients with severe T2DM with the level of glycosylated hemoglobin (HbA1c) exceeding 8.0%. Healthy individuals without aspirin and/or any platelet-affecting agents for at least 7 days prior to the study were also evaluated as the control patients. The institutional review board at the Faculty of Medicine, University of Yamanashi (Yamanashi, Japan), approved the study protocols, and written informed consent was obtained from all the participants.
Collection and Preparation of the Sample for Measurement
Venous blood at fasting stages was carefully collected using Venoject II, a plastic vacutainer (TERUMO, Tokyo, Japan), in which collected venous blood (9 vol) was mixed with 3.13% sodium citrate (1 vol) for anticoagulation. Immediately after blood collection, the sample was centrifuged at 160
Platelet Aggregation
The measurement of platelet aggregation was performed using the light transmittance aggregometry (LTA) method, as described previously. 23 –25 Platelet aggregation was monitored by measuring light transmission with the use of an AG-10 platelet aggregation analyzer (Kowa Co, Ltd, Tokyo, Japan). The AG-10 was calibrated with PRP for zero light transmission and with PPP for 100% light transmission. Platelet aggregation was measured by adding 2 μg/mL collagen (NYCOMED Pharma GmbH, Germany) to PRP preincubated with aspirin. Platelet aggregation was monitored for 7 minutes under constant stirring at 1000 rpm. All the experimental procedures were performed within 2 hours from the initiation of sample blood collection.
Measurement of Collagen-Induced TxB2 Production
Collagen-induced TxB2 production was measured with an enzyme-linked immunosorbent assay (ELISA) kit (Cyman Chemical Company, Ann Arbor, Michigan) according to the manufacture’s instructions. In brief, after monitoring collagen-induced platelet aggregation for 7 minutes, 300 μL acid citrate dextrose (ACD) and 2.8 mmol/L indomethacin were added to each PRP sample in order to completely stop the reaction. The PRP samples were centrifuged at 8000×
Determination of the IC50 Value of Aspirin
The IC50 value of aspirin in each patient with T2DM or healthy individual was calculated using the following formula, which is the triangular approximation of logarithmic model:
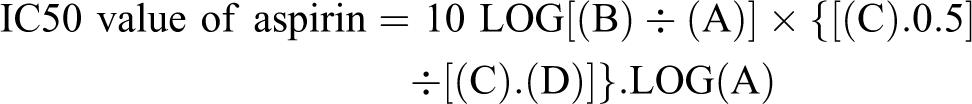
Where, A indicates lower aspirin concentration showing the nearest relative ratio of TxB2 production above 0.5, B indicates higher aspirin concentration showing the nearest relative ratio of TxB2 production below 0.5, C indicates relative ratio of TxB2 production at lower aspirin concentration, and D indicates relative ratio of TxB2 production at higher aspirin concentration.
Each relative ratio of TxB2 production is calculated as the TxB2 production at each aspirin concentration divided by that obtained at 0 μmol/L aspirin (control).
Statistical Analysis
Data were shown as mean ± standard deviation (SD) unless otherwise stated. Categorical variables were expressed as percentages. Statistical analysis was performed using Student t test for continuous variables and the chi-squred (χ2) test for categorical variables. The paired Student t test was applied for the statistical analysis if the data were obtained from the paired patients. On the other hand, if data were obtained from the unpaired patients, F test was initially performed to examine whether the SDs of data obtained from patients were statistically equal. Normal unpaired Student t test would be applied if the SDs were statistically considered to be equal, while Welch method was applied if they were not. All probability values reported are 2-sided, and a value of P less than .05 was considered to be statistically significant.
In addition, regression analysis was performed to explore whether platelet aggregation induced by 30 μmol/L aspirin was possibly correlated with the IC50 values of aspirin and whether diabetic parameters such as HbA1c and glycoalbumin were possibly associated with the IC50 values of aspirin. The authors had full access to the data and took full responsibility for its integrity. Medical records for all participating patients with T2DM and healthy control patients were reviewed.
Results
Baseline Patient Characteristics
Baseline characteristics of patients with T2DM and healthy controls are shown in Table 1. A total of 10 patients with T2DM (age 53.5 ± 16.5 years) and 7 healthy control patients (age 36.9 ± 14.5 years) were enrolled in this study. All the hematological profiles, including platelet counts in PRP, were similar in both the groups. The levels of diabetic parameters, HbA1c (T2DM: 9.6 ± 1.6%, healthy: 5.2 ± 0.2%, P < .005), glycoalbumin (T2DM: 26.1 ± 7.5%, Healthy: 13.4 ± 0.5%, P < .005), and fasting blood sugar (T2DM: 246.7 ± 116.6 mg/mL, Healthy: 75.1 ± 11.0 mg/mL, P < .005) were significantly higher in patients with T2DM when compared with the healthy controls. In addition, the mean values of lipid-related profiles, triglyceride (TG), and low-density lipoprotein cholesterol (LDL-C), exhibited significantly higher levels in patients with T2DM than those observed in the healthy control patients (TG: P < .05, LDL-C: P < .005). The mean value of high-density lipoprotein cholesterol (HDL-C), however, showed no statistically significant difference between the 2 groups.
Baseline Patient Characteristics.a
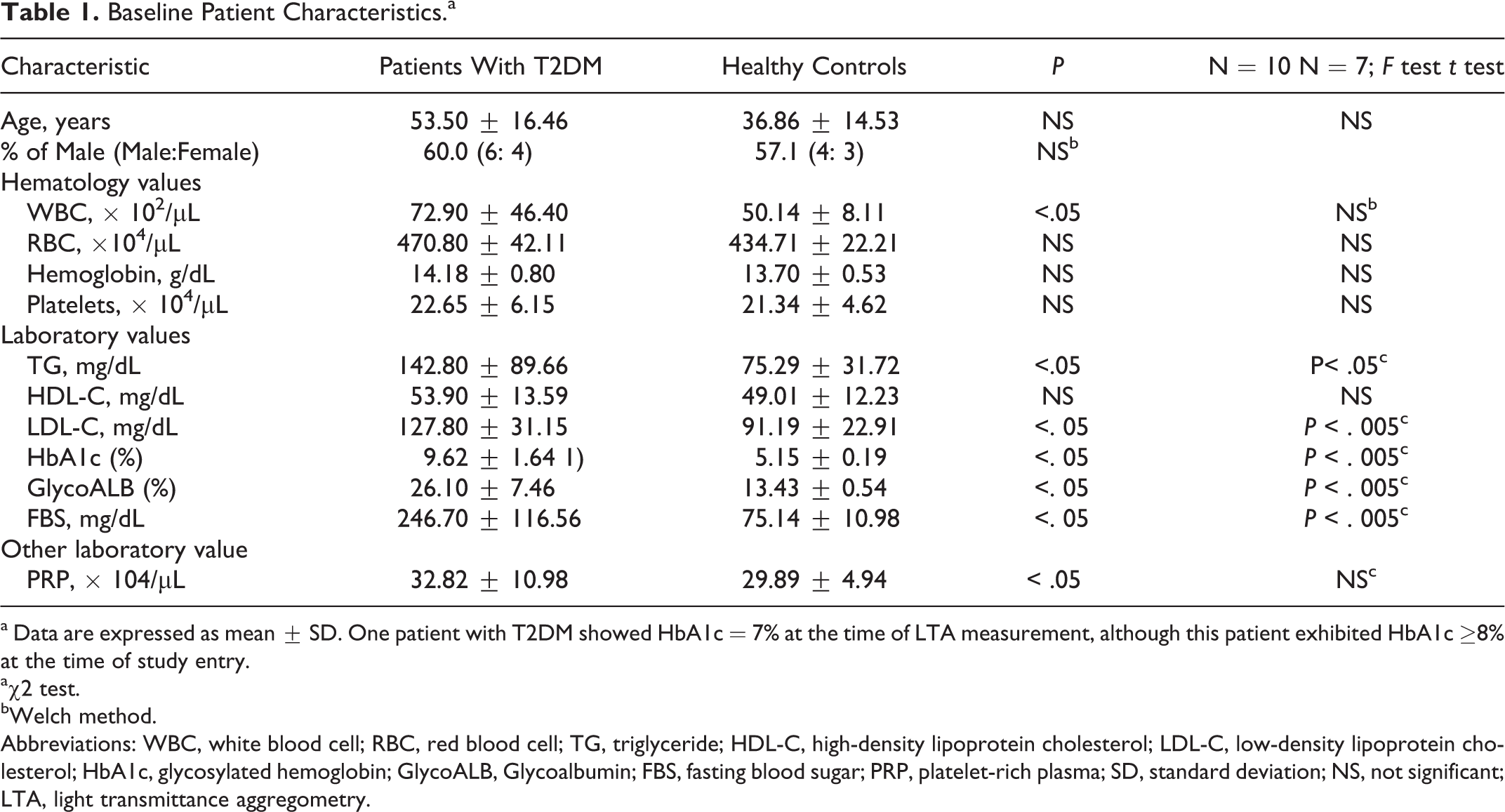
a Data are expressed as mean ± SD. One patient with T2DM showed HbA1c = 7% at the time of LTA measurement, although this patient exhibited HbA1c ≥8% at the time of study entry.
aχ2 test.
bWelch method.
Abbreviations: WBC, white blood cell; RBC, red blood cell; TG, triglyceride; HDL-C, high-density lipoprotein cholesterol; LDL-C, low-density lipoprotein cholesterol; HbA1c, glycosylated hemoglobin; GlycoALB, Glycoalbumin; FBS, fasting blood sugar; PRP, platelet-rich plasma; SD, standard deviation; NS, not significant; LTA, light transmittance aggregometry.
Evaluation of the IC50 Values of Aspirin
The main objective of this study is to evaluate the validity of the Watala’s hypothesis, 22 which states that increased glycation of COX-I may interfere with acetylation of COX-I by aspirin, thereby attenuating the inhibitory effects of aspirin on COX-1. If this hypothesis is correct, the precise assessment of the aspirin potency, such as performed in this study, would yield the IC50 value of aspirin on COX-1 higher in patients with T2DM than in the healthy individuals. This concept can be schematically illustrated in Figure 1, where the IC50 value of aspirin in patient with T2DM shifts to the right (higher level).
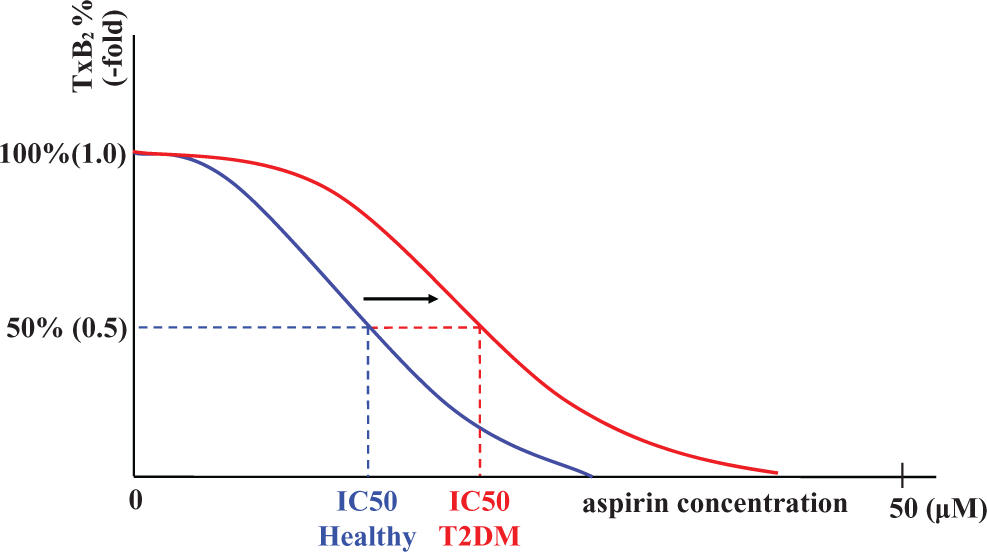
Schematic illustration of the IC50 value of aspirin. If aspirin resistance was present in a T2DM cohort, the IC50 value of aspirin would shift to right (higher concentration). IC50 indicates half maximal inhibitory concentration; T2DM; type 2 diabetis mellitus.
As shown in Figure 2, this study found that aspirin inhibited TxB2 production with virtually the same potency in both patients with T2DM and healthy individuals. There were no significant differences in the IC50 values of aspirin in patients with T2DM (3.16 ± 0.63 μM) and healthy individuals (3.57 ± 0.75 μmol/L; mean ± SD). In addition, the IC50 value of aspirin was not correlated with HbA1c (Figure 3D) or glycoalbumin (Figure 3E) in patients with T2DM.
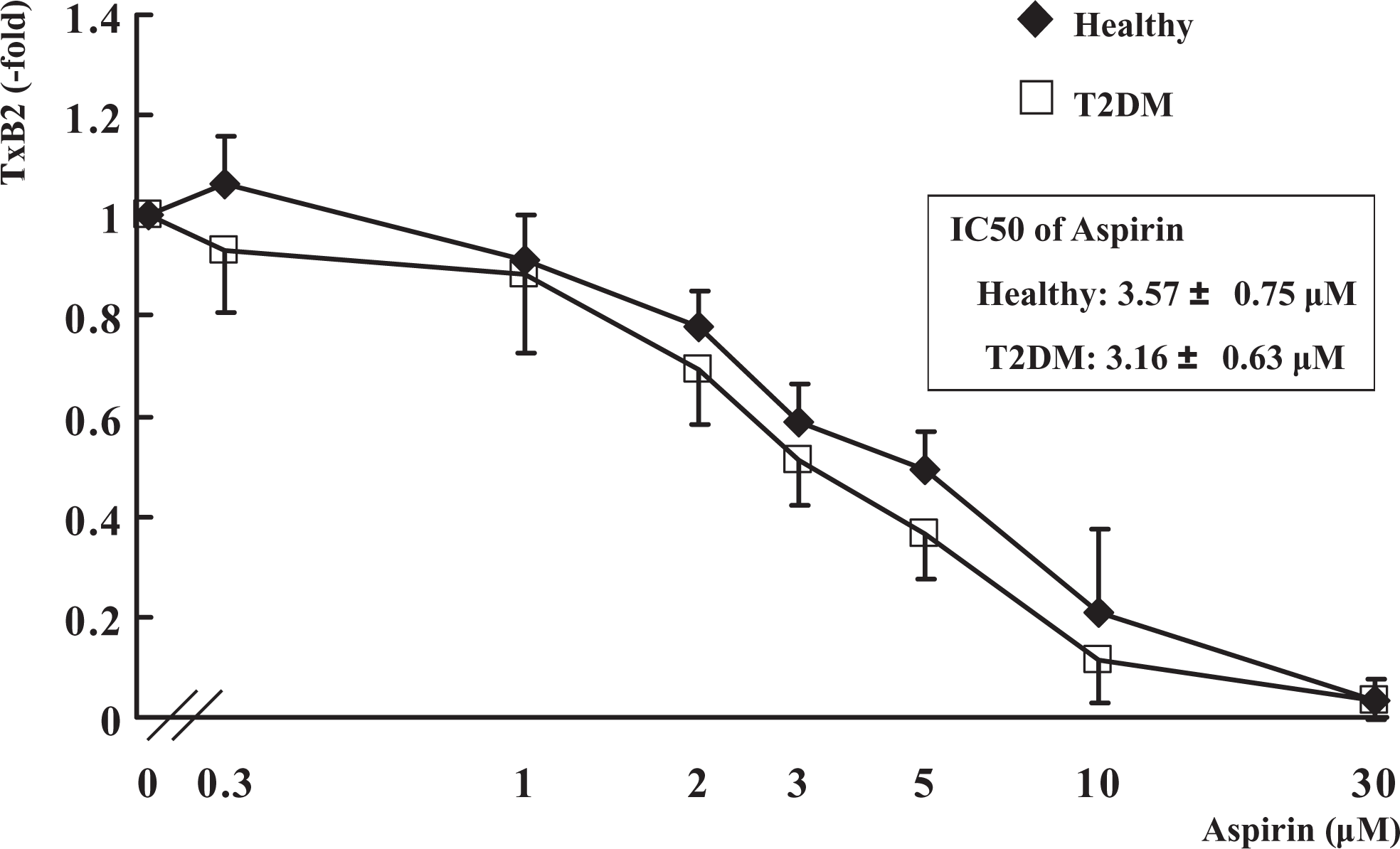
In vitro platelet thromboxane B2 (TxB2) production induc-ed by collagen stimulation. PRP was incubated for 30 minutes with aspirin at the indicated concentrations. After 7-minute stimulation by 2μg/mL collagen, 300 μL acid citrate dextrose (ACD) and 2.8 mmol/L indomethacin were added to each sample to completely stop the reaction. TxB2 production in the supernatant was measured by enzyme-linked immunosorbent assay (ELISA). Data are expressed as mean ± standard deviation (SD).
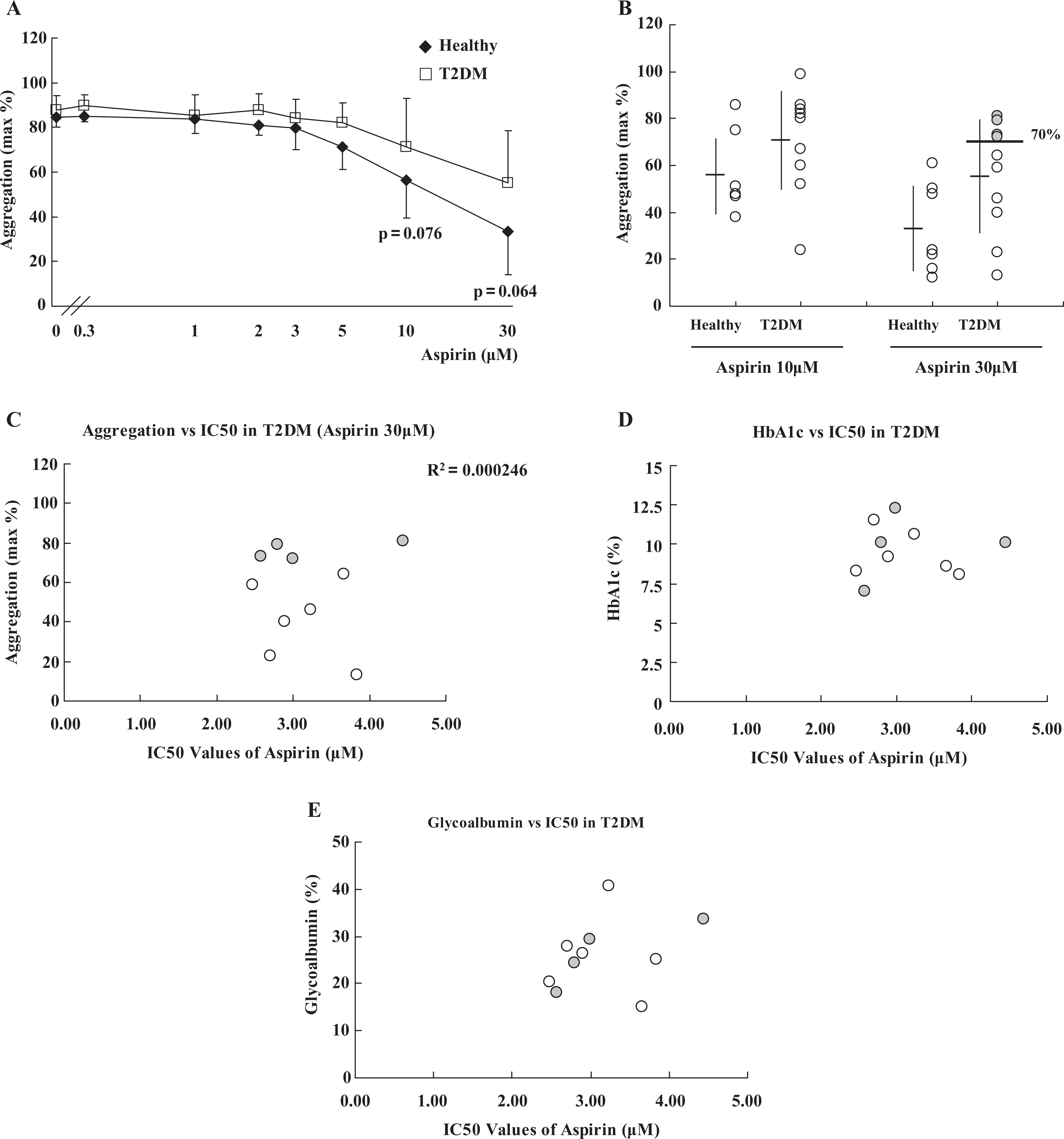
A and B. Maximum platelet aggregation in light transmittance aggregometry (LTA). Platelet-rich plasma (PRP) was stimulated by 2μg/mL collagen for 7 minutes after preincubation with aspirin for 30 minutes at the concentrations indicated. At 10 and 30 μmol/L aspirin, the maximum platelet aggregations in LTA were plotted. Shaded dots denote patients with T2DM exhibiting high residual platelet responsiveness (). Data are expressed as mean ± standard deviation (SD). C, Regression analysis between platelet aggregation and the IC50 value of aspirin in T2DM. Scatterplots were drawn to perform regression analysis exploring whether platelet aggregation was possibly correlated with the IC50 value of aspirin in T2DM patients. Shaded dots denote patients with T2DM exhibiting high residual platelet responsiveness. D and E Regression analysis between the IC50 value of aspirin and glycated hemoglobin (HbA1c) or glycoalbumin in T2DM. Scatterplots were drawn to perform regression analysis, exploring whether the IC50 value of aspirin was possibly correlated with HbA1c. D, glycoalbumin (E) in patients with T2DM . Shaded dots denote patients with T2DM exhibiting high residual platelet responsiveness. IC50 indicates half maximal inhibitory concentration; T2DM; type 2 diabetis mellitus.
Evaluation of the IC50 Value of Aspirin on Platelet Aggregation
With regard to the inhibitory effects of aspirin on platelet aggregation as assessed by LTA, no statistical difference was observed at any aspirin concentration between patients with T2DM and healthy individuals (Figure 3A). However, at the concentrations of 10 and 30 μmol/L aspirin, the T2DM group exhibited a tendency to sustain higher platelet aggregability when compared with that of healthy individuals. This fact implies the presence of aspirin resistance to some extent (10 μmol/L: P = .076, 30 μmol/L: P = .064). The scatterplot of platelet aggregability in the presence of 10 μmol/L or 30 μmol/L was made in order to evaluate the different profile of the aspirin effects on platelet aggregation more closely (Figure 3B). Of the 10 patients with T2DM (shaded dots) 4 showed maximum platelet aggregation exceeding 70% at 30 μmol/L aspirin, while there were no healthy individuals who showed such high platelet responsiveness. It is of interest that TxB2 production was completely inhibited in these 4 patients with T2DM with high residual platelet response in the presence of 30 μmol/L aspirin. It suggests that platelet hyperaggregability is independent of TxA2 production. In addition, regression analysis demonstrates that the IC50 value of aspirin is not correlated with the high residual platelet aggregation in patients with T2DM (shaded dots vs. open dots, Figure 3C). It is noteworthy that the diabetic parameters of these 4 patients with T2DM are not significantly higher than the other patients with T2DM (shaded dots vs. open dots, Figure 3D and E), suggesting that residual platelet response in the presence of aspirin is also independent of protein glycation.
Exploration for the Mechanism of High Residual Platelet Responsiveness
In order to investigate whether one of platelet adenosine diphosphate (ADP) receptors, P2Y12, is engaged in the “high residual platelet responsiveness”, we examined the effect of ARC69931, a specific P2Y12 inhibitor, on aspirin-treated PRP.
Because of the limited volume of clinical samples, we could only evaluate the single concentration of 10 μmol/L aspirin in 5 of the 10 patients with T2DM (including 3 of the 4 patients with “high residual platelet responsiveness”) and all healthy controls. As shown in Figure 4, 1 μmol/L ARC69931 markedly decreased platelet aggregation in all 5 patients with T2DM, suggesting that the P2Y12 pathway plays a role in the “high residual platelet responsiveness.” Maximum platelet aggregation was 81.4 ± 14.1% without ARC69931 and 32.4 ± 20.7% with ARC69931 (P < .005).
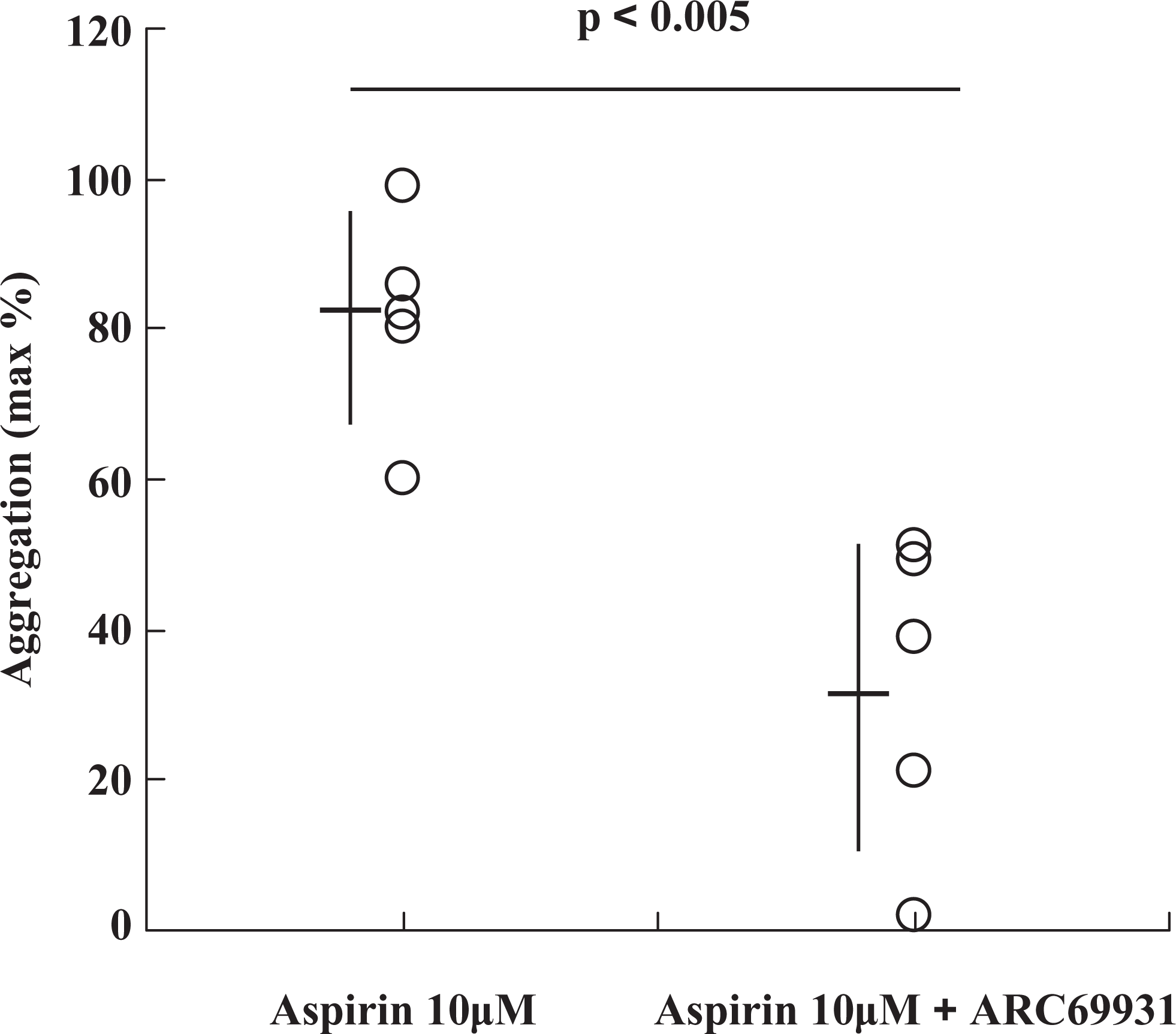
Additive effect of P2Y12 inhibitor on the platelet aggregation in the presence of aspirin in patients with T2DM . The effect of a specific P2Y12 inhibitor, ARC69931, on the collagen-induced platelet aggregation at 10 μmol/L aspirin in 5 patients with T2DM was investigated. PRP was incubated with 10 μmol/L aspirin or 10 μmol/L aspirin + 1 μmol/L ARC69931 for 30 minutes before stimulation of 2μg/mL collagen. Maximum platelet aggregations in LTA were plotted. Data are expressed as mean ± SD. IC50 indicates half-maximal inhibitory concentration; LTA, light transmittance aggregometry; TxB2, thromboxane B2; HbA1c, glycosylated hemoglobin, PRP, platelet-rich plasma;ACD, acid citrate dextrose solution; T2DM, type 2 diabetes mellitus.
Discussion
The limited efficacy of aspirin (aspirin resistance) in a certain population of patients has been known for many years, especially in diabetic patients. Although several mechanisms have been proposed to account for aspirin resistance, it is generally accepted that reduced platelet sensitivity to aspirin, which may be overcome by higher doses, is at least partially responsible for incomplete inhibition of TxB generation. 9,15 Aspirin is considered as a “Pin-Point Bomber” to serine 529 of the platelet COX-1. Aspirin uniformly acetylates serine 529 and inhibits platelet COX-1 activity at very low concentrations. A recent study with Type-1 and Type-2 diabetic patients under aspirin treatment demonstrates that systemic levels of fasting glucose and/or HbA1c are well correlated with in vitro TxB generation. 26 This finding leads to the hypothesis that protein glycation induced by high levels of blood sugar may interfere with acetylation of COX-1 by aspirin, thereby resulting in incomplete inhibition of COX-I by aspirin, that is, aspirin resistance.
In this line of reasoning, 2 articles have been reported. 21,22 According to the first article, hyperglycemia-induced reduction in aspirin sensitivity may be caused by glycation-induced conformational changes in platelet membranes resulting in impaired aspirin entrance. 21 Another article has proposed that less efficient acetylation of platelet proteins by aspirin, possibly including COX-1, occurs under high glucose conditions. 22 In either case, it is conceivable that the extent of reduced sensitivity to aspirin may be partial, and unless an accurate assessment of the TxB generation (the COX-1 activity) such as the IC50 values of aspirin is made, the difference in the COX-I activity cannot be detected. In this study, using various concentrations of aspirin in in vitro TxB generation assays, we evaluated the IC50 values of aspirin on collagen-induced generation of TxB2 in platelets of patients with severe T2DM as well as those of healthy individuals. As far as we know, this is the first study in which the difference in the efficacy of aspirin on COX-1 activity in vitro has been accurately and precisely assessed between the DM and healthy groups. We have found there was no significant difference in the IC50 values of aspirin on COX-1 between those of healthy individuals and patients with severe T2DM (Figure 2).
Since we used intact platelets and assessed the efficacy of aspirin on TxB generation in vitro, the concept of impaired entrance of aspirin due to glycated platelet membranes appears rather remote. 22 Watala et al found that impaired platelet sensitivity to aspirin was related to enhanced platelet protein glycation and reduced incorporation of the acetyl residue of aspirin into platelet proteins in diabetic patients. 22 In addition, the level of protein glycation was inversely correlated with the level of acetylation. 22 Although they have not directly measured the glycation- or aspirin-induced acetylation level of COX-1, they proposed that COX-1 glycation in diabetic patients somehow interferes with acetylation of COX-1 by aspirin, thereby resulting in aspirin resistance. 22 In this study, we found that the efficacy of aspirin on COX-1 is virtually the same between patients with T2DM and healthy individuals, and it is not correlated with HbA1c or glycoalbumin, 2 major markers for protein glycation. Thus, our findings strongly refute their hypothesis that protein glycation affects aspirin efficacy.
Although our findings are not in agreement with this glycation-related hypothesis, Watala findings 22 and ours are not completely at odds with each other. Watala et al did not directly measure TxB generation. 22 They evaluated aspirin efficacy based upon functional studies such as PFA-100TM and arachidonic acid-induced whole blood aggregometry. 22 Based on these functional assays, they suggested that the impaired platelet response to aspirin was related to enhanced platelet protein glycation. We also found out that in 4 of the 10 patients with severe T2DM, collagen-induced platelet aggregation in LTA assay, which is often used to evaluate aspirin efficacy, was only poorly inhibited. Platelet aggregation, however, was uniformly inhibited in all the healthy patients (Figure 3A and 3B). Assuming that platelet proteins are more intensely glycated upon chronic exposure to high glucose levels, as shown by high HbA1c levels, 22 it appears that glycation, even if it occurs on the COX-1 molecule complex, does not affect its sensitivity to aspirin. This may render diabetic platelets more reactive to agonist stimulation through yet unidentified pathways.
It is recently reported that such COX-1-independent “high residual platelet responsiveness” was strongly correlated with poor clinical outcomes. 4,27 Ohmori et al estimated that the contribution of COX-1 in collagen-induced platelet aggregation was approximately 20% at most. 28 Major mechanisms of the “high residual platelet responsiveness” remain unclear. Michelson et al assumed that the “high residual platelet responsiveness” was related to preexistent (before antiplatelet therapy) platelet hyperreactivity to ADP. 29 There are studies indicating that insulin mediates inhibition of the P2Y12 pathway, and that the absence of this inhibition results in the P2Y12 pathway upregulation in patients with T2DM.30–32. In agreement with these reports, we found out that the “high residual platelet responsiveness” was significantly reduced when, in addition to 10 μmol/L aspirin, 1 μmol/L ARC69931, a specific P2Y12 inhibitor, was added to the samples. It suggests that the P2Y12 signaling pathway plays a role in the high platelet responsiveness. Our finding suggests that combination therapy with aspirin and P2Y12 blocker (such as Clopidogrel) may be more effective than aspirin alone, at least for the “high platelet responsiveness” individuals. In CURE trial, Clopidogrel (75 mg/day followed by 300 mg/day) and aspirin (75-325 mg/day) were administered in patients with acute coronary syndromes (ACS) without ST-segment elevation. 33 This study suggested the beneficial effects of the dual antiplatelet therapy of Clopidogrel and aspirin on those patients, although caution should be paid to such dual therapies since dual antiplatelet therapy overall may increase bleeding complications. 34 It has been recently reported that platelet function monitoring with or without adjusting antiplatelet dose showed no significant improvements in clinical outcomes. 35,36 Although these results may argue against the need for platelet function monitoring / antiplatelet dose adjustment, it is too premature to draw conclusion based on a limited number of studies.
In the present study we have demonstrated that the IC50 values of aspirin on COX-1 in patients with T2DM were similar to as in healthy individuals. These results indicate that the hypothesis that high blood glucose levels with resultant glycation of lysine residues of COX-1 interfere with aspirin acetylation of the COX-1 active site is unlikely. 37 Although aspirin almost completely inhibited the TxA2 /TxB2 production both in patients with T2DM and the healthy patients, persistent platelet aggregation induced by collagen despite high aspirin doses was observed only in the T2DM group. Such “high residual platelet responsiveness”, which might lead to thrombotic events in patients with T2DM, was corrected by adding a P2Y12 receptor inhibitor. It implies that P2Y12 inhibitors, such as Clopidogrel, in addition to aspirin would be beneficial for these cases. Our study has a limitation of power due to the small sample size. Further large-scale study should be implemented to clearly elucidate the definition of “aspirin resistance” in patients with T2DM.
Footnotes
Acknowledgments
We would like to thank the following colleagues in the study for their cooperation, expertise and excellent technical assistance: K. Suzuki-Inoue MD PhD, O. Inoue MD PhD, N. Sugiyama, K. Ohshimo, C. Komatsu, J. Nakagomi, H. Nakazawa, M. Hara, M. Shibata, M. Osada, M. Ohta, F. Kazama, Wuxun Jin MD PhD, Andrew Kelliher RPh MS MBA, George Alexis RPh MS, Harold Sparr RPh PharmD, Linda S Restino RN MS, Ronald Goren MD PhD, and Alla Gulyansky MD PhD.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.