
Abstract
The cirrhosis population represents a unique subset of patients who are at risk for both bleeding and developing venous thromboembolic events (VTEs). It has been commonly misunderstood that these patients are naturally protected from thrombosis by deficiencies in coagulation factors. As a result, the cirrhosis population is often falsely perceived to be “autoanticoagulated.” However, the concept of “autoanticoagulation” conferring protection from thrombosis is a misnomer. While patients with cirrhosis may have a bleeding predisposition, not uncommonly they also experience thrombotic events. The concern for this increased bleeding risk often makes anticoagulation a difficult choice. Prophylactic and therapeutic management of VTE in patients with cirrhosis is a difficult clinical problem with the lack of clear established guidelines. The elucidation of laboratory and/or clinical predictors of VTE will be useful in this setting. This review serves to examine VTE and the use of anticoagulation in the cirrhosis population.
Introduction
The prevalence of chronic liver disease and cirrhosis in the United States is estimated to be over 5 000 000, as reported by the American College of Gastroenterology study in 1998. 1 Cirrhosis is characterized by fibrotic remodeling of the liver architecture into abnormal nodularity with fibrous septations. Fibrotic obliteration of the sinusoidal spaces, together with local compressive effects leads to increased pressure in the portal venous system. These changes impair liver function and are partially characterized by both decreased synthetic capability and clearance of bioactive and toxic products. As a result of these defects, patients with cirrhosis often accumulate excessive activated coagulation factor proteins, mediators of fibrinolysis, increased platelet consumption, sequestration, reduced platelet production, and acquired platelet dysfunction. Patients with this malady are often admitted to the hospital for management of complications which include hepatic encephalopathy, ascites, spontaneous bacterial peritonitis, hepatorenal syndrome, and venous thrombosis. Hemorrhagic complications also occur, such as capillary-type bleeding of mucosal surface, puncture sites, petechiae/purpurae/ecchymoses, epistaxis, menorrhagia, and, most notably, variceal bleeding. Prolonged international normalized ratio (INR) and partial thromboplastin time (PTT) reflecting impaired hepatic synthetic functions are hallmarks of cirrhosis. With progressive cirrhosis, reduction in vitamin K-dependent factors starting with factor VII in the early stages followed by factors II, X, and IX in the later stages contribute to INR prolongation. The PTT is also prolonged from decreased synthesis of factors XI and XII along with the deficiencies in vitamin K-dependent factors II, IX, and X. When taken together with concomitant thrombocytopenia and clinical features of hemorrhagic tendency, patients with cirrhosis are often perceived to be “autoanticoagulated” and hence protected from venous thromboembolic events (VTEs). However, accumulating evidence has not only mitigated the role of these coagulation defects in, for example, variceal bleeding but also found little evidence to suggest protection against VTE. This is in part supported by a similar decrease in natural anticoagulants (antithrombin, protein S, and protein C) that is a consequence of the global decrease in hepatic synthetic function. Therefore, the term “autoanticoagulation” is a misnomer. 2,3 Venous thromboembolic events are herein defined as either deep venous thrombosis (DVT) and/or pulmonary embolism (PE). Other forms of venous thrombosis affecting the portal, splanchnic, and hepatic veins which are also encountered in cirrhosis are beyond the scope of this article. This review serves to examine VTE and the use of anticoagulation in the cirrhosis population with acquired coagulation abnormalities.
Incidence of VTE in Cirrhosis
Evidence suggests that the coagulopathies frequently encountered in patients with cirrhosis do not confer adequate protection from VTE. The incidence of VTE in hospitalized patients with cirrhosis is estimated to be 0.5% to 1.9% but has been reported to be as high as 6.3%. 4 –8 Hospitalized patients with cirrhosis without predisposing comorbidities (eg, neoplasm, congestive heart disease, and chronic renal failure) have similar risks of VTE as compared to patients without cirrhosis. 5 Sogaard et al conducted a large nationwide case–control study using population-based data from the Danish national registry looking at 99 444 cases of VTE and 496 872 controls to demonstrate an increased relative risk (RR) for VTE (RR 1.74: 95% confidence interval [CI], 1.54-1.95) in patients with underlying cirrhosis after adjusting for predisposing factors such as neoplasm, fractures, trauma, surgery, pregnancy, and Charlson comorbidity index. 9 This study also established that there exists greater RR for DVT (RR 2.02: 95% CI, 1.78-2.31) than PE (RR 1.41: 95% CI, 1.20-1.65) in the population studied. This is consistent with the previous studies which have found that most PEs originate from DVT of the lower extremities and further adds to the validity of this study’s conclusions. 10 Of particular interest, when the investigators restricted the analysis only to patients with unprovoked VTE, there appeared to be an increased RR (2.06: 95% CI, 1.79-2.38). Another population-based study carried out in the United States by Wu et al looking at over 649 000 cirrhosis cases and 575 000 controls also reported an increased VTE risk in hospitalized patients with cirrhosis up to the age of 45 after adjusting for age, gender, race, and comorbidity. 7 Taken together, these data suggest that some patients with cirrhosis are not protected from VTE. In addition, they may have blood clotting abnormalities that may also place them at risk for bleeding. Unfortunately, none of the studies cited above specifically investigated thrombotic risk categorized by the presence or absence of specific coagulopathies, thrombocytopenia, or platelet dysfunction.
Predictors of VTE in Cirrhosis
Laboratory/Clinical Parameters
An interesting case–control study by Northup et al examining more than 21 000 patients with cirrhosis having 113 cases of VTE determined that an elevated INR does not confer protection from VTE. 4 However, it did identify decreased serum albumin (odds ratio [OR] 0.25: 95% CI, 0.10-0.56, P < .001) as an independent predictor of VTE. Gulley et al comparing hospitalized cirrhosis patients with matched noncirrhosis controls also identified albumin as a predictor of VTE (OR 0.47: 95% CI, 0.23-0.93, P < .05). 5 A more recent study by Dabbagh et al stratifying the severity of coagulopathy in cirrhosis using INR quartiles of 1.4/1.7/2.2 did not demonstrate significant difference in VTE incidence. 8 These results are supported by the knowledge that despite impaired synthesis of clotting factors involved in the forward reactions of coagulation, liver synthetic impairment also leads to reduced generation of the natural inhibitors of these same reactions (eg, proteins C, S, and antithrombin). This impaired biosynthesis is accompanied by an impaired hepatic clearance of activated clotting factors and regulators of fibrinolysis that are not quantified by INR measurement. 11 Of particular relevance, since the INR was developed as a method of minimizing differences in prothrombin time (PT) measurement among patients receiving vitamin K antagonists (VKAs) caused by the use of thromboplastin agents of varying sensitivity, it does not necessarily reflect either severity or adverse risk in patients with liver disease with same fidelity as with patients receiving VKA. 12,13 Patients on VKA when compared to those with cirrhosis have different coagulation defects. 14 Furthermore, the traditional method of deriving the INR has demonstrated wide variability in patients with cirrhosis between different centers, thereby creating bias in the allocation of livers for transplantation that is reliant upon the model for end-stage liver disease (MELD) score which incorporates the INR. 13,15 This has led to the recent proposal for an alternative calibration scale specific to cirrhosis. 14 The use of this proposed cirrhosis-specific INR calibration scale in predicting the risk of VTE is unexplored. Unlike the INR however, albumin has been shown to be a more accurate surrogate for VTE in this patient population. The relationship between low albumin and VTE mirrors that of nephrotic syndrome where the hypoalbuminemic state has a significant association with VTE. 16 Perhaps, they may share similar mechanisms that remain incompletely understood.
Other common laboratory variables that have been investigated including aspartate aminotransferase, alanine aminotransferase, total bilirubin, platelet count, creatinine (Cr), and blood urea nitrogen have not been shown to be clinically useful in predicting VTE. The use of composite clinicolaboratory assessment as defined by the Child-Pugh score taking into account total bilirubin, INR, albumin, the presence of ascites, and encephalopathy also failed to demonstrate a statistically significant difference between VTE incidence in Child A versus Child B/C cirrhosis. 5 One critique against the use of clinical parameters is the presence of interobserver variability expected from the clinical interpretation of the grade of ascites and encephalopathy. Other composite models such as the MELD score involving the use of Cr, INR, and total bilirubin have also been studied and expectedly failed to demonstrate a predictive role, given their nonassociation individually with univariate analysis. 4
Demographic/Comorbid Risk Factors
In the previously discussed Sogaard et al study, a greater risk for unprovoked VTE was reported to exist in patients aged <55 years (RR 3.58: 95% CI, 2.62-4.88). 9 However, a subgroup analysis of patients with cirrhosis and concomitant hepatocellular carcinoma did not find an increase in VTE risk. Similarly, in the nationwide population-based study in the United States, Wu et al found a 25% increased VTE risk in cirrhosis patients aged <45 years when compared to those aged ≥45 years. 7 Data from both of these studies demonstrated an increased risk in the younger cirrhosis population. When examining VTE risk in cases of cirrhosis and comparing them with controls from other disease states, the estimated risk for VTE in cases of cirrhosis yielded 1.9% while the corresponding risks in noncirrhosis controls with a known neoplasm, chronic renal failure, or congestive heart disease were estimated to be 6.1%, 7.0%, and 7.8%, respectively. 5 Therefore, the risk for VTE in cirrhosis is lower than that in other diseases known to predispose to VTE. Further studies investigating whether concurrent cirrhosis has a risk-modifying effect for VTE in patients with known neoplasm, chronic renal failure, or congestive heart disease are awaited.
Coagulation/Fibrinolysis Status in Cirrhosis
Coagulation
Broadly, hemostasis is established by 2 concurrent processes involving the formation of tissue factor (TF):activated factor VII (aFVII) complex, and the formation of platelet plug involving the binding of von Willebrand factor (vWF) to platelets and collagen which also further serves as a scaffold for TF:aFVII complex activity. The TF:aFVII complex, through activation of factor X and with aFV as a cofactor, is responsible for the conversion of prothrombin to thrombin. Activated factor X and thrombin participate in a positive feedback loop by converting the cofactors FV and FVIII to their activated form. These activated cofactors then act as amplifying agents that enhance the downstream interaction between FIX and FX and activated aFVII and FX. This amplification results in “burst” of thrombin production and enhanced fibrin formation. The extra thrombin burst also feedback to factor XI which then stimulates the production of additional aFIX which also contributes to more thrombin production and its downstream consequences.
Abnormalities occurring in cirrhosis include thrombocytopenia, reduction of procoagulant FII, FV, FVII, FIX, FX, FXI, and FXII which participate in the forward reactions of coagulation, and the natural inhibitors antithrombin, protein C, and protein S. Typically FVIII activity and its carrier vWF are increased in patients with liver disease, since they are synthesized at sites external to the liver parenchyma. Fibrinogen is synthesized in the liver and its production may be either depressed or the molecule may be dysfunctional in patients with liver disease. Decreased fibrinogen activity is usually seen in the late stages of cirrhosis. Although dysfibrinogenemia has been described in patients with liver disease, it is rarely of clinical significance contributing neither to bleeding nor to thrombotic risk. 17 However, it must be noted that decreased fibrinogen or dysfibrinogenemia may influence PT and PTT and thrombin time measurement independent of other coagulation factors. Many patients have concomitant thrombocytopenia from splenic sequestration and decreased synthesis of thrombopoietin-regulating platelet production. 18 They also experience qualitative platelet defects with decreased interaction with subendothelium and impaired platelet aggregation. 19,20 Although vWF levels are elevated in cirrhosis, there is disruption of the usual vWF function. 21 The significance of these findings is uncertain; however, some postulate that the higher than normal levels of vWF may compensate for decreased platelet numbers and dysfunctional vWF activity. 22 Although this postulation seems tenuous, it remains a possible explanation for the maintenance of hemostatic function in patients with cirrhosis with thrombocytopenia. Also, to some extent, this postulation is supported by clinical experience with restoration of platelet function with either cryoprecipitate (which are rich in vWF) or desmopressin infusions (which induce the release of vWF from Weibel-Palade bodies). ADAMTS 13 (also known as vWF-cleaving protein) is a protease synthesized in the liver. In noncirrhosis disease states characterized by deficiencies in ADAMTS 13 activity, vWF analysis reveals the presence of ultrahigh-molecular-weight (UHMW) multimers of vWF. 23 The UHMW multimers provide excessive receptor binding sites for specific glycoproteins on the platelet surface and enhance platelet adhesion and aggregation. ADAMTS 13 activity has been reported to be depressed in some patients with cirrhosis but not in others. 22,24 This may provide a rationale for the maintenance of improved or normal hemostasis seen in patients with cirrhosis with thrombocytopenia and increased, but dysfunctional, vWF.
The PT and, by extension, the INR for the issues previously described, are poor methods for monitoring coagulation status in cirrhosis especially since they do not take into account the concomitant decrease or increase in coagulation factors or natural inhibitors in cirrhosis. Recent studies investigating the endogenous thrombin potential (ETP) of plasma samples from individuals with and without cirrhosis have confirmed that plasma samples from patients with cirrhosis characteristically have the capacity to produce appropriate if not excessive amounts of thrombin in relation to similar samples from patients without cirrhosis. 25 –27 However, severe thrombocytopenic states in cirrhosis can limit thrombin generation. 28 Although the significance of excessive ETP remains uncertain, mounting data support the contention that it is associated with hypercoagulable disorders and that this method of testing has global implications since it takes into account the effect of elements involved in the forward reactions, reverse reactions, and natural inhibitors of coagulation. 29,30
Another interesting observation is related to thrombomodulin (TM) resistance that has been described in patients with cirrhosis. Plasma obtained from patients with worsening cirrhosis indicates the occurrence of a reciprocal increase in resistance to TM that is associated with the diminishing ability to activate protein C. 31 Thrombomodulin is a protein found both on phospholipid surfaces (eg, vascular endothelium) and in plasma. Thrombomodulin binds thrombin to inhibit coagulation by promoting the activation of protein C. 32 Activated protein C is responsible for slowing the forward reactions in coagulation by inhibiting the activities of FVIII and FV, the cofactors responsible for the amplification of these pathways. It has been postulated that this phenomenon may be due to the coagulation imbalance favoring the excess of procoagulation factors. When taking together the relationship between worsening cirrhosis and increased FVIII levels that is accompanied by decreased protein C levels, it has been reasoned that the imbalance of these 2 potent mediators of coagulation induces a procoagulation shift that could account for the enhanced ability of plasma from patients with cirrhosis to generate thrombin and account for an overall prothrombotic state. 31
Fibrinolysis
The coagulation system is a finely balanced mechanism between the forward, reverse, and inhibitory pathways. Without this balance, there would exist excessive predisposition toward either thrombosis or bleeding. The reverse reactions of coagulation include the natural breakdown of fibrin mesh by the process of fibrinolysis.
Fibrin is initially broken down by plasmin, which is activated from its inactive form, plasminogen, through the action of tissue plasminogen activator (tPA). Fibrinolysis is driven by tPA and regulated by inhibitors including plasminogen activator inhibitors (PAIs-1) and antiplasmin (AP). Thrombin-activatable fibrinolysis inhibitor (TAFI), a protein that inhibits plasmin, is activated shortly after formation of thrombin from its precursor protein prothrombin and acts as an immediate downregulator of fibrinolysis. Both TAFI and AP are synthesized in the liver. Their plasma levels have been reported to be reduced in patients with cirrhosis. 17 Similarly, plasminogen, the precursor protein of plasmin, is also synthesized in the liver and its levels are depressed in cirrhosis. 17 In contrast, tPA is largely derived from vascular endothelial cells and PAI-1 is synthesized at multiple sites other than the liver. 33,34 However, the clearance of both tPA and PAI-1 (after binding to tPA) are hepatic dependent. 35 With the exception of acute hepatic failure, plasma levels of tPA in cirrhosis are increased in relation to PAI-1. 36 This difference may be due to either the distinct sites of synthesis for the 2 proteins or a difference in plasma clearance between tPA and tPA:PAI-1 complex. Profibrinolytic changes include decreased levels of TAFI and AP that is accompanied by increased levels of tPA. Antifibrinolytic changes include decreased plasminogen and an increased PAI-1. The net effects of these changes have been suggested by some to be hyperfibrinolytic, especially in subgroup of patients with more advanced cirrhosis and may contribute to the bleeding risk in patients with cirrhosis. However, this concept of a cirrhosis-associated hyperfibrinolytic state remains contested (Table 1). 17,37 –40
Coagulation Imbalance in Cirrhosis.

Abbreviations: AP, antiplasmin; PAI-1, plasminogen activator inhibitor 1; TAFI, thrombin-activatable fibrinolysis inhibitor; tPA, tissue plasminogen activator.
Prophylaxis of VTE/Treatment in Cirrhosis
The latest 2012 American College of Chest Physicians guidelines for VTE prophylaxis and treatment do not provide a specific recommendation on management of patients with cirrhosis. 41 Presumptively, the cirrhosis population may fall under the category of patients with increased risk of hemorrhage. For prophylaxis, mechanical compression devices are recommended in patients with a high risk of hemorrhage. In those with established VTE, the placement of inferior vena cava filter is recommended in situations where anticoagulation is not a reasonable alternative.
Kanaan et al conducted a systematic review looking at anticoagulation prophylaxis in unselected hospitalized medical patients and found an 1.7% absolute increase in minor bleeding with no increased risk for major bleeding and an 1.36% reduction in absolute risk for DVT with a number needed to treat 74 to prevent 1 episode of DVT. 42 We currently have no data to estimate these risks in patients with cirrhosis. Although pharmacological anticoagulation prophylaxis is the goal standard for VTE prevention, its use in patients with cirrhosis plus coagulopathy has been controversial, given the risk of hemorrhage and the belief that an elevated INR confers some protection. The prevalence of nonprophylaxis in hospitalized cirrhosis patients has been quoted to be as high as 75%. 8,43 The efficacy of VTE prophylaxis in cirrhosis has been poorly characterized, but the previously introduced study by Northup et al (with 113 cases of VTE) reported that 7% and 14% of patients experienced VTE while receiving either anticoagulation or mechanical prophylaxis, respectively. 4 The combination of the low incidence of VTE and the large proportion of patients with cirrhosis who do not receive pharmacologic prophylaxis in published studies have posed difficulties in evaluating the efficacy of VTE prophylaxis. 8 The frequency of hemorrhage-related morbidity from pharmacological prophylaxis was not reported. Data from the population-based study by Wu et al estimated a 2-fold increase in mortality and length of hospitalization among patients with cirrhosis. They recommended VTE prophylaxis in cirrhosis patients aged <45 years and an individual case-based consideration in patients aged ≥45 years. 7 These recommendations seem tenuous since they do not to take into account individualized cirrhosis-associated bleeding risk or the occurrence of concomitant coagulopathy.
Safety of Anticoagulation
Clinical data pertaining to the safety of VTE pharmacological prevention and treatment in the cirrhosis population are meager. Garcia-Fuster et al reported the anticoagulation experience in 17 patients with VTE and cirrhosis. 6 All patients received low-molecular-weight heparin (LMWH) with 6 patients being bridged to VKA after 1 week of LMWH therapy. The authors reported significant hemorrhagic complications in 14 (83%) patients, with 6 (35%) being severe as defined by the requirement for blood transfusion. Severe hemorrhage was however noted to be more frequently associated with VKA use (83.3%). In the absence of more robust anticoagulation data in cirrhosis patients with VTE, inferential data from small studies of cirrhosis patients with portal/splanchnic vein thrombosis (PVT/SVT) and other comorbidities requiring anticoagulation can be mined for pertinent information. The prospective cohort study by Bechmann et al examining the prophylactic use of LMWH in the form of enoxaparin 40 mg/d in 75 patients (Child B—50.7%, Child C—37.3%, and Child A—12%) recorded hemorrhagic complications in 5 (7%) recipients. 44 This same study examined the use of enoxaparin 1 mg/kg twice daily therapeutic dose in 9 patients (Child B—55.6%, Child C—11.1%, and Child A—33.3%) and reported hemorrhagic complications in 2 (22%) recipients. Observed hemorrhagic complications consisted of either bleeding esophageal varices or hypertensive gastropathy with no mortality. Although the authors reported similar hemorrhagic rates in a comparable cirrhosis population not on anticoagulation therapy, it must be noted that this study was underpowered, a fact that draws into question its validity. 45,46 Most recently, Delgado et al shared their anticoagulation experience using either LMWH or VKA in 55 patients with PVT and reported hemorrhagic complications in 10 (18%) patients. 47 Of these, 5 patients experienced variceal bleeding with the remainder consisting of mucocutaneous typed bleeding such as oral, vaginal, nonvariceal gastrointestinal, and surgical wound bleeding. A platelet count of <50 × 109/L was found to be associated with increased hemorrhage and while not statistically significant, the use of VKA was more commonly observed to cause hemorrhage. Amitrano et al examined the treatment of PVT with enoxaparin 200 units/kg per d (2 mg/kg per d) for at least 6 months in 28 patients and reported only 7% hemorrhagic complications from hypertensive gastropathy. 48 All patients were able to complete at least 6 months of anticoagulation without interruption from hemorrhage. On closer examination of these 28 patients, 46.4% had Child B/C cirrhosis and 50% were noted to have presented with bleeding varices. Those presenting with bleeding varices commenced anticoagulation only after endoscopic ligation and β-blocker prophylaxis. Senzolo et al analyzed their anticoagulation experience in 26 selected patients from a pool of 38 patients with PVT and based upon their findings of only 1 case of hemorrhagic complication, it was suggested by the authors that LMWH anticoagulation can be safely used in cirrhosis after careful evaluation and treatment of any varices by successful variceal banding therapy. 49 In their report, 12 patients did not commence anticoagulation either because of suboptimal results from endoscopic banding therapy or because of stable cavernous transformation with reperfusion of intrahepatic portal vein. The smaller study by Francoz et al consisting of 19 patients with cirrhosis with SVT receiving nadroparin 5700 IU/d for at least 5 days (followed by VKA with a target INR of 2-3) recorded only 1 case of variceal bleeding. 50 This bleeding episode was iatrogenic, resulting from a postligation bleeding ulcer that occurred as a complication of prophylactic variceal banding. At the recent 2011 American Association for the Study of Liver Diseases (AASLD) conference, Villa et al presented data from a randomized control study demonstrating the safety and efficacy of enoxaparin 40 mg/d in the prevention of PVT among patients with cirrhosis. 51 No hemorrhagic events were recorded in the treated patients. Importantly, an improved survival outcome was seen with the treatment versus placebo group. One caveat to note is that while many of these studies have the ability to provide a crude gauge on the type and frequency of hemorrhagic complications associated with patients undergoing anticoagulation, the accurate interpretation of the direct role of anticoagulation on hemorrhagic outcomes in cirrhosis remains difficult since it is unknown whether these hemorrhagic episodes are directly instigated by anticoagulation or they may represent the natural history of cirrhosis that is complicated by PVT/SVT.
Portal hypertension and bacterial infections are 2 important risk factors for hemorrhage in patients with cirrhosis. When referring to hemorrhage encountered in cirrhosis, a distinction must be made between a capillary-type and variceal bleeding. Capillary-type bleeds present as epistaxis, petechiae, and low volume oozing bleed from puncture sites (eg, dental extractions, postprocedural taps, and drains). Conversely, esophageal variceal bleeding is a more sinister complication with brisk blood loss occurring in approximately 30% of the cirrhosis population and accounts for 80% to 90% of bleeding episodes in these patients. Unfortunately, variceal bleeding in patients with cirrhosis is associated with up to 30% mortality at the first episode and carries a 70% recurrence rate. The 1-year survival is estimated to be from 32% to 80%. 52 These data indicate that variceal bleeding is an ominous feature. While defective coagulation in decompensated liver failure has been established to be associated with nonvariceal capillary-type bleeding, there has been paucity of data linking it with variceal hemorrhage. 53 The primary etiology for bleeding varices is from increased portal pressure from portal hypertension. 53 –55 Accordingly, β-blocker prophylaxis and shunt procedures aimed at mitigating portal hypertension have been the mainstay treatment. Attempts to reduce bleeding risk in the liver transplant setting by infusing large amounts of fresh frozen plasma (FFP) to correct prolonged clotting times have been largely ineffective and has often led to increased intravascular volume with resultant increased blood loss. 56 In murine models of cirrhosis with acute blood loss, volume repletion aimed at compensating for blood loss has been shown to produce elevation of portal venous pressure over baseline and resulted in greater rebleeding and mortality. 57 , 58 Accordingly, the AASLD practice guidelines in 2007 has recommended caution against aggressive volume resuscitation in the setting of acute variceal hemorrhage. 59 Besides highlighting the importance of hemodynamic alteration in variceal bleeding, the aggressive correction of coagulation defect in cirrhosis has little significance in preventing blood loss. A large multicenter trial investigating the utility of aFVII in active variceal bleed has also failed to establish a beneficial effect on a composite primary end point consisting of control of active bleed, prevention of rebleeding, and 5-day mortality. 60 Furthermore, the INR has been demonstrated to correlate poorly with hemorrhagic complications in cirrhosis. 61 –63 Therefore, there is insufficient evidence to support the role of clotting factors derangement as a major contributor to severe hemorrhagic episodes in this setting.
As previously mentioned, another important factor to consider is the presence of bacterial infections. 64,65 Upper gastrointestinal bleeding is associated with bacterial infection in 33% to 66% of patients with cirrhosis and the utility of antibiotic prophylaxis has been proven to decrease rebleeding from acute variceal hemorrhage. 66,67 It has been established that cirrhosis patients with bacterial infections may experience the untoward effects of endogenous heparinoids (as determined by heparinase-modified thromboelastography testing) which prolong the thrombin time and increase bleeding risk in a manner similar to that seen in patients receiving heparin products. 68 Because of this, the elevated risk for hemorrhagic complications in patients with superimposed infection must be considered when deciding on anticoagulation (Table 2).
Hemorrhagic Complications Associated With Anticoagulation in Cirrhosis.
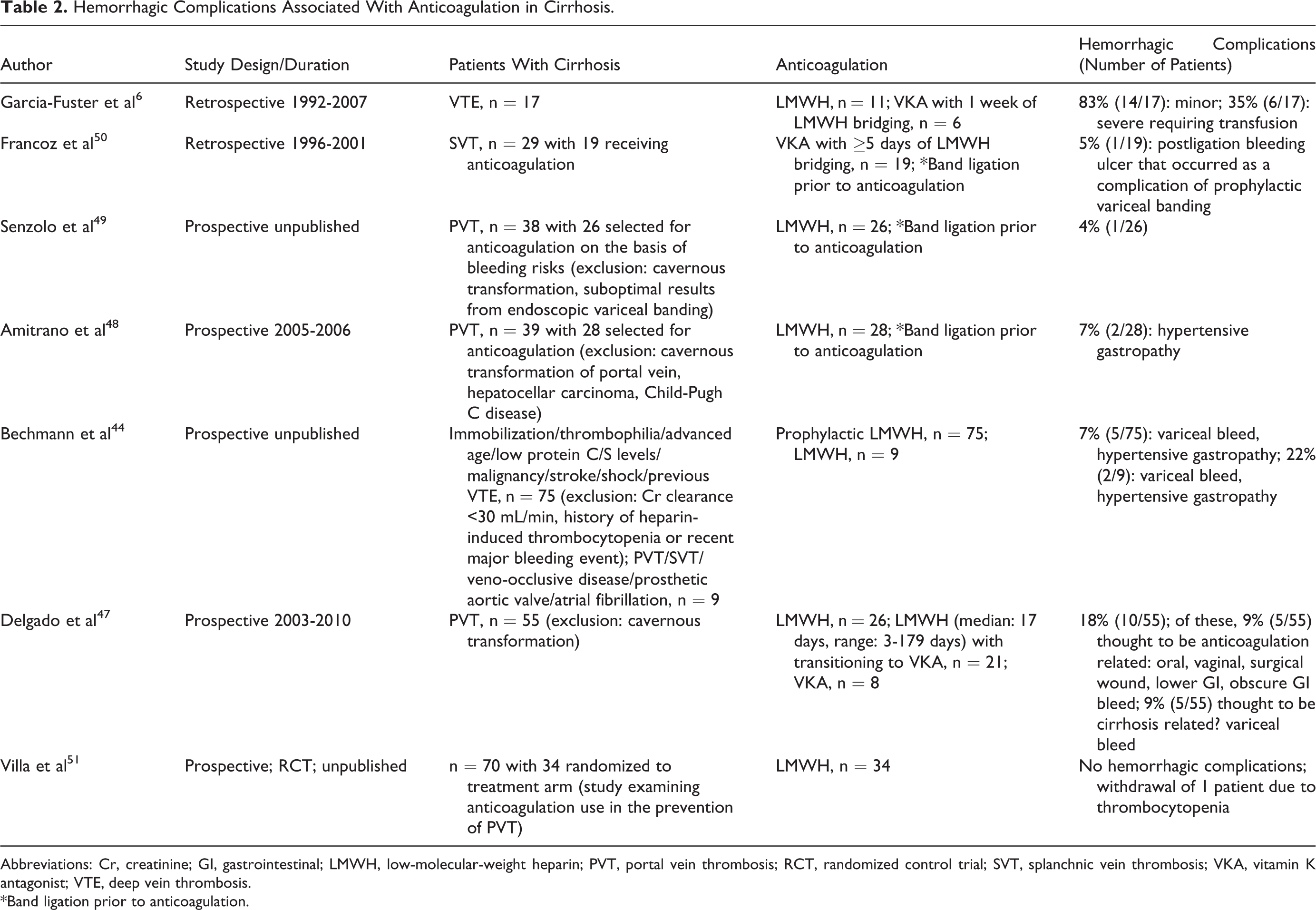
Abbreviations: Cr, creatinine; GI, gastrointestinal; LMWH, low-molecular-weight heparin; PVT, portal vein thrombosis; RCT, randomized control trial; SVT, splanchnic vein thrombosis; VKA, vitamin K antagonist; VTE, deep vein thrombosis. *Band ligation prior to anticoagulation.
Pathogenesis of Cirrhosis: Role of Thrombosis
Thrombosis has been implicated in cirrhosis progression. An early study by Wanless et al examining explanted liver specimens suggested that PVT and hepatic vein thrombosis encountered in liver cirrhosis contribute to intimal fibrosis and cirrhosis progression. 69 This observation may be explained by local ischemia leading to hypoxia-mediated mechanisms involving expression of collagen type 1 by hepatic stellate cells and expression of angiogenic factors. 70 Also, thrombin mediates signaling through protease-activated receptors (PARs) on hepatic stellate cells to potentiate tissue remodeling and fibrogenesis, leading to cirrhosis. The application of thrombin inhibitors and PARs antagonist has been shown to protect against liver fibrosis in murine models. 71,72 Furthermore, clinical evidence in patients with hepatitis C with FV Leiden mutation indicates that these patients experience a more rapid progression of liver fibrosis, whereas patients with hepatitis C with hemophilia experience a milder course. 73,74 Along with this data, available evidence on animals has suggested anticoagulation with either LMWH or VKA may prove to be beneficial in slowing cirrhosis progression in humans. 75,76
Conclusions
Undoubtedly, liver cirrhosis encompasses a unique coagulopathy where patients are not only prone to hemorrhage but also remain susceptible to thrombosis, possibly from imbalances of pro-/anticoagulation factors. Difficulties exist in identifying which patients will bleed or develop clot. This is likely the reason why pharmacological VTE prevention has not been universally accepted in this population. There is absence of well-validated clinicolaboratory markers to guide the safe and appropriate use of anticoagulation in cirrhosis. Although the INR has been a useful test in determining the adequacy of anticoagulation and bleeding tendency in patients receiving VKA, its use as a measure of anticoagulation status in patients with cirrhosis is flawed. Unlike recipients of VKA with intact liver function, the functional status of the extrinsic coagulation pathway (routinely measured with INR) alone is not sufficient to proclaim true “autoanticoagulation” status in cirrhosis. This rationale is supported by the fact that the liver is responsible for the generation of other antithrombotic factors and for the clearance of both activated thrombotic factors and factors associated with fibrinolysis. Moreover, attempts to correct elevated INR in variceal bleeding by administration of FFP or aFVII have not been beneficial.
Other commonly available laboratory tests utilized to provide a brief guide to the state of coagulation and fibrinolysis pathway include
Footnotes
Acknowledgment
The authors acknowledge Dr Gladwin, MT, for providing recommendations and for critical appraisal of the manuscript.
Author’s Note
Y.Z.J. drafted and revised the manuscript. S.R.E. reviewed, edited, and revised the manuscript. N.E.M. and C.K.A. reviewed and edited the manuscript.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported in part by the National Institutes of Health through Grant Numbers UL1 RR024153 and UL1TR000005.