
Abstract
Myeloproliferative neoplasms (MPNs) are clonal hematopoietic stem cell disorders and are characterized by advanced proliferation and survival advantage. These abnormalities are considered to derive from the point mutation in exon 14 of the Janus kinase 2 genes (JAK2 V617F). The thrombosis rate and the high prevalence of JAK2V617F in patients with MPN suggest that there is an association between the 2 in MPN. Apart from the mutation, other variables are documented to cause endothelial dysfunction and involve in thrombotic tendency. Endothelial progenitor cells differentiated from hematopoietic stem cell in patients with JAK2V617F MPN play an indispensable role in initiating and modulating neoangiogenesis. Although a risk-oriented therapeutic approach has been applied to MPN treatments, the further study on pathogenesis of MPN may provide more novel preventions and therapies for MPN.
Breakpoint cluster region (BCR)/Abelson (ABL)-negative classical myeloproliferative neoplasms (MPNs), characterized by the overproduction of cells without obvious blockage in maturation from one or more myeloid lineages, likely originate at the level of hematopoietic stem cells. 1 –4 These related conditions have predominant erythroid lineage or megakaryocytic/megakaryocytic–granulocytic lineage. 2 According to the 2008 World Health Organization classification system for hematologic malignancies, MPN include 4 “classic” disorders: chronic myelogenous leukemia, polycythemia vera (PV), essential thrombocythemia (ET), and primary myelofibrosis (PMF). The differences in their genetic profiles lead to their phenotypic diversity.
Abnormalities of the JAK2 pathway are considered as the primary molecular lesion implicated in the pathology of MPN, and JAK2V617F has been one of the criteria for the diagnosis of MPN. 2,5,6 In about 70% of patients with MPN, many studies have described a mutation that valine substitutes with phenylalanine at position 617 (V617F) of the JAK2 protein, in the pseudokinase domain (JH2). 2,3 It is believed that the V617F mutation alleviates some of the negative autoregulation on the kinase domain generated by the JH2 at the molecular level. 1,3 The JAK2V617F mutation leads to a gain of function resulting from constitutive activation of tyrosine kinase-dependent cellular signaling pathways, particularly the JAK-signal transducer and activator of transcription (JAK-STAT) pathway. 1,5,7 This mutation likely provides progenitor cells with both a proliferation and a survival advantage. 3,8
Etiological Factor and Pathogenesis of Thrombosis in MPN
Thrombotic disorders have been most extensively investigated in PV and ET and will be the focus of the current work. Data related to myelofibrosis will not be discussed in detail in this review in consideration of limited present researches on it. Mechanisms involved in thrombotic tendency have been recently ascribed in part attributed to the quantitative and functional abnormalities of erythrocyte, thrombocyte, and leukocyte, as well as the molecular hallmark of MPN, JAK2V617F mutation. 9
Main causes of morbidity and mortality in patients with MPN are usually attributed to leukemic transformation, infection, bleeding, portal hypertension, as well as vascular complications. 10 The rate of thrombosis occurred in PV and ET can reach 50%. 11,12 A recent large study of patients with PMF also showed a similar cardiovascular thrombotic risk to that in ET. 13 The percentage of deaths from thrombosis was reported up to 13.4% to 70% and 27.3% to 30.7% in patients with PV and ET, respectively. 11,12 This issue raised the interest in the causes of thrombosis and how to prevent them. And up to now, many possible factors have been suggested to contribute to the thrombotic formation in different researches. But the detailed correlation between these factors and thrombosis is still controversial, with one study suggesting an association, whereas others objecting to it. In this review, we will discuss several of them in detail.
Erythrocytosis and Hyperviscosity
Patients with PV with an increased hematocrit (HCT) are at an augmented thrombotic risk due to the hyperviscosity associated with the high HCT. Earlier clinical studies showed an incidence of thrombosis reaching 60% in patients with PV with a HCT above 60%. 14 Hyperviscosity can trigger thrombosis through enhancing erythrocytes aggregation, disturbing blood flow, facilitating thrombocyte aggregation and activation, and prompting thrombocyte and leukocyte interaction with the vessel wall. 15,16
Thrombocytosis
It is generally considered that increased platelet count contributes to the pathogenesis of thrombotic process. Thrombocytosis increases dynamic interactions between platelets. Microvascular complications can be ameliorated when the platelet count in patients with MPN returns to the normal range. 17 In patients with ET, of note, bleeding is more likely to occur than thrombosis along with extreme thrombocytosis, especially the platelet count is greater than 150 × 109/L. 18 This phenomenon can be attributed to the qualitative abnormalities of von Willebrand factor (vWF). It has been observed that an increased platelet count can perturb vWF activity (collagen-binding activity and istocetin cofactor activity) rather than the vWF count. Possible pathogenesis is speculated that increased platelet bind to vWF, facilitate platelet activation and aggregation, and promote vWF multimers proteolysis, lead to increased consumption and loss of vWF large multimers and the consequent formation of anomalous vWF activity. 19 –21
Leukocytosis and Leukocyte Activation
Leukocytosis (uncertain high threshold count) is a risk factor for thrombosis in patients with MPN, in whom qualitative abnormalities can occur, leading to increased leukocyte activation promoting prothrombotic changes. In patients with MPN, neutrophil abnormalities correlate with increase in the plasma levels of prothrombotic markers (
JAK2 V617F Mutation
Since 2005, the JAK2V617F mutation has been described in 65% to 97% of patients with PV, in approximately 23% to 75% in ET and 30% to 50% in PMF. 1 It has been hypothesized that association may exist between JAK2 mutation and thrombosis in MPN. 27
De Stefano et al carried out a retrospective cohort study including 224 patients with ET; they reported that the presence of the JAK2 V617F mutation increased the relative risk of overall thrombosis by 45%. The risk was significant for both arterial and venous thromboses. However, after having grouped the patients according to the site of venous thrombosis, the risk associated with the JAK2 V617F mutation was significantly increased for venous thromboses of unusual sites (ie, splanchnic or cerebral vein thrombosis). 28
However, Iványi et al found that the incidence of thrombosis ensued in JAK2-negative MPN cases did not differ significantly from the JAK2-positive patients and suggested that the presence or absence of JAK2 mutation in the development of thrombosis has no predictive value in patients with MPN. 29 These discrepancies can be accounted for sample size, study design, therapy, and the length of observation period. Therefore, a large, multicenter study may be necessary for resolving the problem. But thus far, the implications of JAK2V617F mutation on thrombosis risk are unclear (Figure 1).
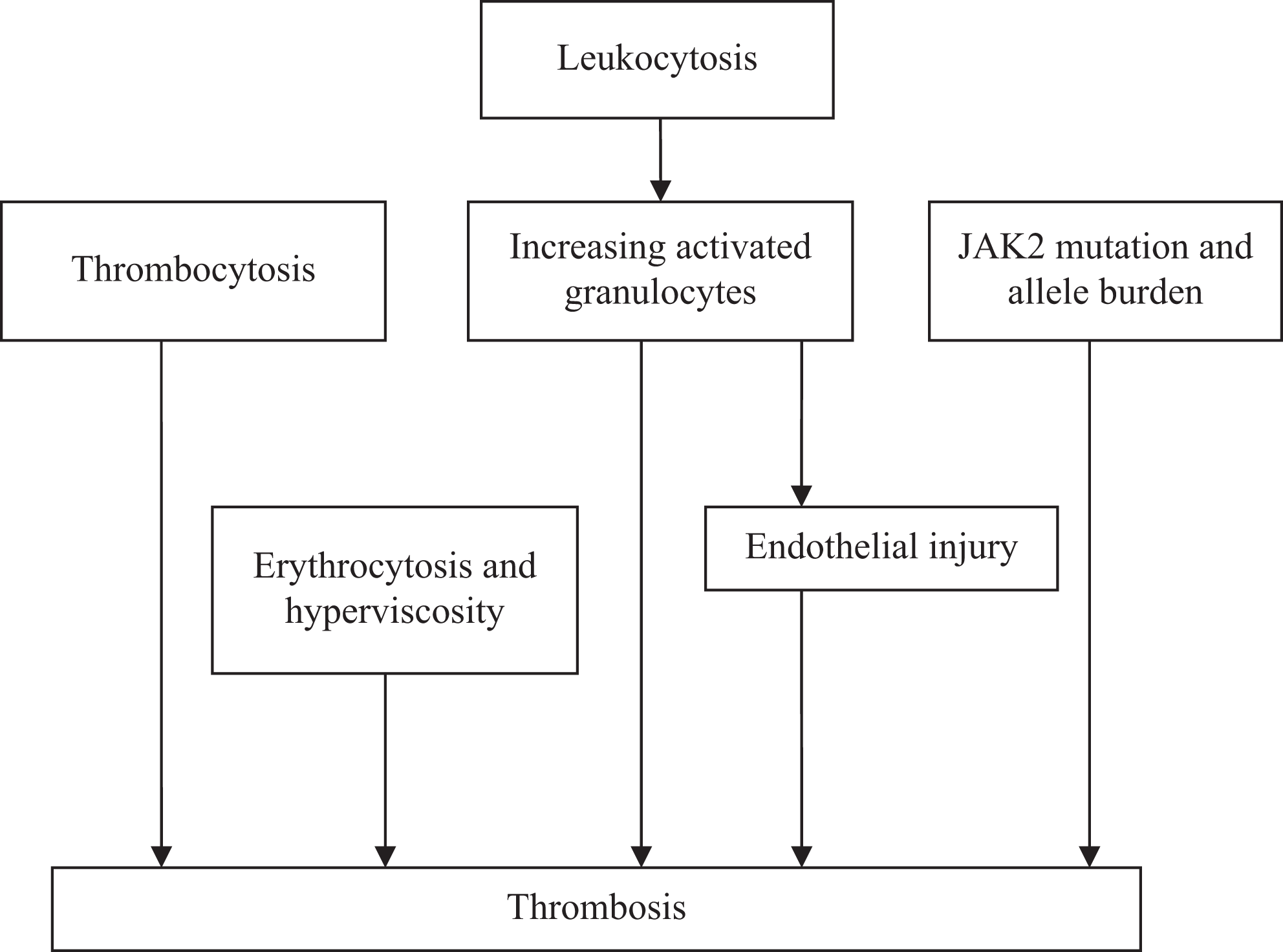
Possible etiological factors for thrombosis.
The Possible Molecular Biology Mechanism in Patients With MPN With JAK2V617F Leading to Thrombosis
JAK2 Mutation
Identification of the JAK2V617F mutation in 2005 boosted basic and clinical research in BCR/ABL-negative MPN.
According to the assessment of the JAK2-mutated allele burden in patients with MPN, higher values are found in PV and the lower are in ET, the so-called “gene-dosage hypothesis” are suggested, and it speculates the mutated allele burden would be the most important contributor to MPN phenotype. 30,31 In the same direction, homozygosity for the JAK2 mutation is frequent in PV and rare in ET. Anand et al demonstrated in vivo that JAK2 mutations do not alter hematopoietic stem and progenitor cell compartment size or in vitro behavior but generate expansion of later myeloid differentiation compartments, where homozygous expression of the mutation confers an added proliferative advantage at the single-cell level. 32
One research suggests the JAK2V617F mutation burden was positively correlated with levels of hemoglobin, HCT, and leukocyte in PV and ET samples. 31 All the high value of HCT and leukocyte are risk factors for thrombosis. Antonioli et al assessed 260 patients with ET and showed increasing mutant allele load correlated with higher frequency of arterial thrombosis at diagnosis. 33
Regina et al demonstrates the existence of a specific association between unexplained splanchnic vein thrombosis and the JAK2 mutation through analyzing the blood samples collected from 44 patients who had developed portal or hepaticthrombosis and supports latent MPN can favor the development of idiopathic splanchnic vein thrombosis. 34 A recent study showed that platelets from patients with the JAK2 V617F mutation more easily expressed P-selectin 35 and this process may contribute to the pathogenesis of splanchnic vein thrombosis through combining with the low flow existing in the portal vein system.
Search for additional mutations in patients with Ph-negative MPN led to the discovery of the JAK2 exon 12 mutations, which occur exclusively in PV, and mutations of the MPL gene in about 5% of ET and 10% of patients with PMF. 36,37 The W515L mutation of the MPL gene confers cytokine-independent growth and thrombopoietin hypersensitivity and results in constitutive activation of the JAK2-STAT pathway. 36 More recent studies have revealed a number of novel mutations in patients with BCR/ABL-negative MPN (eg, TET2, EZH2, ASXL1, etc). However, none of them was MPN specific, displayed mutual exclusivity, or could be traced back to a common ancestral clone.
In summary, the molecular genetics of BCR/ABL-negative MPN is much more complex than initially believed after the discovery of the JAK2V617F mutation. Up to now, it remains unknown which mutation is the initiating genetic event. The repertoire of other mutations is increasing but our understanding of their pathogenic role is limited by their lack of restriction to specific disease phenotypes.
Endothelial Progenitor Cell
The BCR/ABL-negative MPN shows a high incidence of vascular complications and an endothelial cell (EC) dysfunction has been evidenced in patients with PV. 38
Actually, during the embryonic development, hematopoietic cells and ECs derive from a common precursor called hemangioblast, which arises from mesoderm. 39 Teofili et al not only showed that the same molecular signatures are detectable in endothelial progenitors and in the hematopoietic clone but also found the presence of JAK2V617F mutation into endothelial progenitors is associated with the hyperphosphorylation of STAT-3 and STAT-5. 40 Previous studies also have evidenced similar results. Sozer et al not only reported the presence of JAK2V617F mutation in the liver endothelial cells of patients with PV with Budd-Chiari syndrome but also demonstrated that CD34 cells isolated from peripheral blood of patients with PV or PMF with JAK2V617F are capable of generating both wild-type and JAK2-mutated endothelial-like cells when transplanted into nonobese diabetic/severe combined immunodeficient mice. 41 In addition, Yoder et al reported that a minority of endothelial colony-forming cells (E-CFCs) derived from a patient with vascular thrombosis and subsequently developing JAK2V617F PV, carried the JAK2V617F mutation. 42 However, Piaggio et al firmly excluded that E-CFCs obtained from patients with MPN (including patients with chronic myeloid leukemia) harbored the disease-specific molecular clonality marker (JAK2V617F mutation or BCR/ABL rearrangement). 43 Of note, the endothelial progenitors have identified JAK2V617F mutation or specific chromosome alterations only derived from the hematopoietic lineage (the so-called colony-forming unit-endothelial cells [CFU-ECs]), 42,44 whereas the true E-CFCs do not harbor genetic abnormalities. 42 This may partly contribute to the discrepancies in above different researches.
The former results from Teofili et al add evidence to the existence of a common hematopoietic and endothelial bi-lineage progenitor also in BCR/ABL-negative MPN. From a clinical point of view, the latter discovery will promote more endothelial progenitors adhering to normal mononuclear cells and, therefore, increase the incidence rate of thrombosis.
In general population, the decreased number of circulating endothelial progenitor cells (EPCs) predicts future cardiovascular events, and it is has been hypothesized that this mechanism could account for the increased thrombotic risk in patients with PV. 38 Teofili et al found a normal range of true circulating endothelial progenitors in their patients with PV and ET, but an increased number in PMF for patients with history of thrombosis, the CFU-EC level was significantly lower than in healthy participants. 40 The increased level of E-CFCs may explain the lower thrombosis rate in patients with PMF.
Teofili et al found that the clonality and JAK2 mutation appeared in E-CFCs were associated with a thrombotic proficiency. Interestingly, these patients have high JAK2 allele burden and high leukocyte count at diagnosis 40 ; these 2 factors have been demonstrated to associate with an increased thrombotic risk. They also show that endothelial cells with JAK2V617F mutation abnormally activates the JAK/STAT pathway, which is an important regulator of the response of endothelial cells to injury and increases their proficiency to adhere to normal mononuclear cells. 40 The above discoveries may support the hypothesis that the pathogenesis of thrombosis in MPN may result from an EPC dysfunction.
As far as we know, vascular ECs participate in both vascular repair and reendothelialization of damaged blood vessels. And the fact that whether ECs function well or not is a major factor for vascular endothelium repair or thrombosis. It needs to further study whether the function of ECs differentiated from JAK2V617F-postive EPCs has any defects and whether these defects influence the repair of injured vascular endothelium. The understanding of the problem would reveal the mechanisms involved in the formation of thrombosis in patients with MPN from the aspect of defective differentiation of EPCs or even hemangioblast and provide novel ideas and theoretical basis for antithrombotic therapy in patients with MPN.
In addition, investigational thrombotic risk factors include inherited thrombophilia, panmyelosis, and traditional cardiovascular risk factors (eg, smoking, hypertension, overweight, high cholesterol, etc), which are still need to validate in prospective studies.
Anyway, in view of contrary conclusions drawn from different studies, further researches are necessary for the association of thrombosis in PV, ET, PMF, and these risk factors, respectively. And these disease-related risk factors may play different roles in these 3 MPNs due to the concurrence of 2 or more risk factors. The clear relationship of these risk factors and MPN may provide a new guideline for the risk stratification and therapy of MPN.
Antithrobmotic Therapy of MPN
Given that thrombosis represents the leading cause of death in patients with PV and ET, the primary goal of treatment is to prevent thrombosis at the time of diagnosis, or even much earlier, especially for those patients with PV/ET with high-risk factors including older than 60 years or a prior history of vascular events. 45 Patients with PV or ET are classified in a “high-risk” or “low-risk” category according to their age and previous history of thrombosis. Current managements based on the risk stratification of these MPNs are recommended by the consensus of experts. 46,47 But with the discovery of other possible risk factors, it has posed a challenge to the current risk stratification system (Table 1).
Risk Stratification and Treatment for PV/ET.
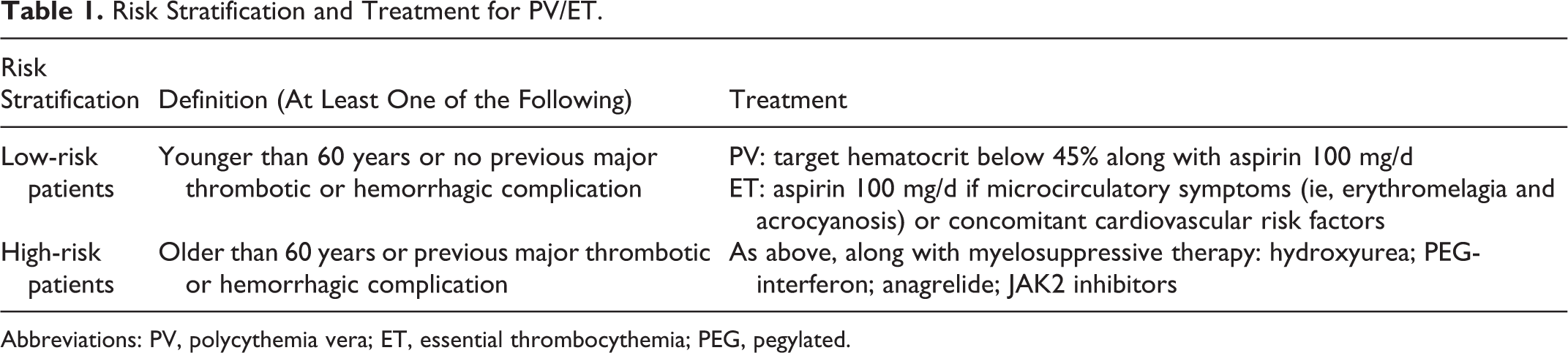
Abbreviations: PV, polycythemia vera; ET, essential thrombocythemia; PEG, pegylated.
Low-Risk Patients (Age Less Than 60 Years, No Previous Thrombosis)
Multicenter random studies have showed that low-dose aspirin (≤100 mg/d) combined with control of erythrocytosis therapy has markedly decreased the occurrence of vascular events. 48,49 For all patients with PV with no contraindication to this treatment, phlebotomies and aspirin constitute the optimal management. 50
Phlebotomy
More than 20 years ago, the Polycythemia Vera Study Group demonstrated that patients with PV treated in the phlebotomy arm had a better overall median survival (13.9 years) than the other 2 arms (chlorambucil 8.9 years, radiophosphorus 11.8 years) due to a reduced incidence of acute leukemia and other malignancies. 51 In contrast, thrombotic events were more frequent in the phlebotomy arm in which the target HCT was set at 45% on the basis of a small, retrospective study. 14 Given the association, several studies tried to probe the correction between HCT values and thrombosis, but the results are still unclear. Despite that, it still seems a wise way to normalize and maintain HCT values less than 45%.
Low-Dose Aspirin
The European Collaborative Low-dose Aspirin has assessed the efficacy and safety of low-dose aspirin (≤100 mg daily) in PV cases in double-blind, placebo-controlled, randomized clinical trial. In this study, data analysis showed a significant reduction of a primary combined end point including cardiovascular death, nonfatal myocardial infarction, nonfatal stroke, and major venous thromboembolism (VTE) comparing to the control. 52
Aspirin, 100 mg daily, has been found to control microvascular symptoms in ET. However, the conflicting opinions expressed in national guidance and consensus documents challenges the application of standard dose aspirin as a primary prophylaxis for low-risk ET. 53 In these patients, the thrombohemorrhagic complications may be present and the role of aspirin has traditionally been controversial. Alvarez-Larran et al has questioned the benefit of low-dose aspirin in patients with low-risk ET and suggested that antiplatelet therapy reduces the incidence of venous thrombosis in JAK2-positive patients and the rate of arterial thrombosis in patients with associated cardiovascular risk factors. No benefit was found from low-dose aspirin in JAK2-negative or in patients without cardiovascular risk factors in whom only observation may be an adequate option. 54
High-Risk Patients (Older Than 60 Years, Previous Thrombosis)
For high-thrombotic risk participants, administration of cytoreductive therapy together with aspirin is recommended. 50 Apart from advanced age and prothrombotic history, poor tolerance to or high need of phlebotomy, symptomatic or progressive splenomegaly, severe disease-related symptoms, platelet counts greater than 1.5 × 109/L or progressive leukocytosis are other indications for cytoreduction. Three main agents—hydroxyurea, anagrelide, and interferon all can lead to cytoreduction. 48,49
Hydroxyurea
As an antimetabolite, hydroxyurea prevents DNA synthesis and reduces thrombosis in PV and ET without enhancing the risk of leukemia transformation. 55 In 1 trial, 114 patients were randomized to 2 groups, one group (n = 56 cases) with long-term hydroxyurea treatment and another group (n = 58 cases) with no cytoreductive treatment. During a median follow-up of 27 months, 2 thromboses were recorded in the hydroxyurea-treated group compared to 14 in the control group. Notably, the antithrombotic effect of hydroxyurea may be a result of panmyelosuppression, qualitative changes in leukocytes, decreased expression of endothelial adhesion molecules, and increased nitric oxide generation. 56
Interferon α
In MPN, interferon α (IFN-α) was considered to suppress the proliferation of hematopoietic progenitors, to inhibit bone marrow fibroblast progenitor cells, and to antagonize the action of platelet-derived growth factor, transforming growth factor-β and other cytokines, thought to be involved in the development of myelofibrosis. 57 Pegylated forms of IFN-α is more convenient than IFN-α, as pegylated IFN-α allows for weekly administration. This improves compliance and is possibly a more effective therapy. A phase 2 study has shown that the percentage of the mutated allele JAK2V617F was reduced from a mean of 45% to a mean of 22.5% after 12 months of treatment with pegylated IFN-α-2a (PEG-IFN-α2a), with no evidence for a plateau being achieved. The tolerability of PEG-IFN-α2a at 90 mg weekly was optimal. 57,58 However, whether this drug is more efficacious than hydroxyurea in reducing the rate of vascular events remains to be demonstrated.
In patients with PV who proved intolerant or refractory to hydroxyurea or IFN-α, one can substitute the 2 drugs with each other as the first-line therapy.
Anagrelide
Anagrelide, a member of the imidazoquinazolin compounds, has a potential to downregulate platelet activity without a leukemogenic effect. This appeared to be an alternative option to hydroxyurea for reducing platelet counts in younger patients with ET. In a randomized clinical trial including 809 patients with ET, treatment with anagrelide was compared with treatment with hydroxyurea (along with aspirin in both groups). 59 Compared with patients on hydroxyurea, patients in the anagrelide arm showed an increased rate of arterial thrombosis, major bleeding, and myelofibrotic transformation but a decreased incidence of venous thrombosis. Therapy with anagrelide, but not with hydroxyurea, was also associated with progressive anemia and increased bone marrow fibrosis. 60 Anagrelide is recommended as a second-line therapy in ET cases refractory or intolerant to hydroxyurea. 47
The use of low-dose aspirin in patients with ET is not yet as substantiated as in patients with PV and its efficacy has been demonstrated in small studies only. 61 On the other hand, in patients with ET, more and more thromboxane biosynthesis and selective aspirin efficacy in microvascular complications are discovered, which may provide a biological platform for its practice. 58
It is still not yet defined how long an optimal therapy should be maintained when treating thrombotic event in patients with MPN. Additional large epidemiological studies on this subject are necessary. De Stefano et al reported that for patients with PV or ET, advanced age (>60 years), and thrombophilia in younger patients are the main risk factors for the recurrences of thrombotic events. 62 As MPN is an irreversible acquired risk factor for VTE disease, anticoagulants are recommended for an unlimited period after one thrombotic episode.
Novel Drugs
As the alterations in JAK-STAT pathway plays a central role in the clinical manifestations of MPN, targeting JAK2 appears an appealing therapeutic goal. 48,49 Several drugs displaying differential inhibitory activity against JAK family members and on other receptor kinases, such as fibroblast growth factor receptor, platelet-derived growth factor receptor, and FMS-like tyrosine kinase 3, are currently under clinical development. 8 Safety, efficacy, and toxicity data on novel inhibitors such as Ruxolitinib (INCB018424), Lestaurtinib (CEP701), and TG101348 are still limited. There is still a long way to go before they are available for clinical use.
Clinical trials thus far have demonstrated that several of these selective JAK2 inhibitors could meaningfully reduce MPN-associated constitutional symptoms. The JAK2 inhibitors provide durable benefits and symptomatic benefits not seen with previous generations of nontargeted therapies. Currently, Ruxolitinib, as an adenosine triphosphate-competitive orally bioavailable small-molecule inhibitor of JAK1 and JAK2 kinases, has been for the first time approved to the treatment of patients with PMF and post-PV or -ET myelofibrosis due to its significant beneficial effects on spleen size reduction and symptom improvement.
Other Therapies for MPN
Individuals with MF traditionally have a worse natural history. This is thought to be the most severe MPN because PMF presents with progressive marrow fibrosis, leukoerythroblastosis, and splenomegaly. 63 The main treatments include hematopoietic stem cell transplantation, traditional drugs (androgens, erythropoietin, thalidomide, and hydroxyurea), splenectomy, or splenic radiotherapy and new drug therapy (hypomethylation, antifibrotic drugs, immunomodulatory drugs, and inhibitors of JAK2).
Conclusion
The discovery of the JAK2V617F mutation in 2005 strengthened the link between PV, ET, and PMF and provided, for the first time, the rationale for the pathogenesis of thrombosis and development of targeted therapies for these MPNs. JAK2 mutation, endothelial cells and overproduction of one or more myeloid lineages have been suggested to contribute to the thrombosis in MPN. As a consequence of the development of whole genome assays, an increasing number of mutations have been observed in BCR/ABL-negative MPN, these mutations involved in genes modifying epigenetic regulation add more complexity to the pathogenesis of MPN. The main limit of current studies resides mainly in its retrospective nature other than small sample size, study design and so on, and only prospective and large investigations may definitely confirm present observations. Contrary views consequently result from these limits. Future studies could aim at identifying detail mechanisms of the signaling pathways which regulate gene expression and promote survival, proliferation, and differentiation of committed myeloid progenitors, as well as identify mutations present in MPN in order to better understand the molecular cause, evolution of the clone, and the effects of the different mutations. All these may provide new therapies for patients with MPN and a novel method to understand cancer development result from stepwise mechanisms in humans.
Footnotes
Acknowledgment
The authors would like to thank Prof Man-Chiu Poon for critical review of the manuscript.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship and/or publication of this article: Tianjin Municipal Science and Technology Commission (11JCZDJC18600 and 09ZCZDSF03800) and Ministry of Science and Technology of China ((2012AA02A211and 2010DFB30270). The authors would like to thank Prof. Man-Chiu Poon for critical review of the manuscript.