
Abstract
Intra-abdominal thrombosis is a complication of paroxysmal nocturnal hemoglobinuria (PNH). There is scarcity of data on cases presenting with thrombosis in whom PNH is the predisposing factor. We assessed the role of PNH defect in 81 patients with intra-abdominal thrombosis, 44 patients of Budd Chiari syndrome and 37 patients of extra hepatic venous obstruction. Flowcytometry with glycosylphosphatidyl inositol-anchored proteins (GPI-AP)-CD55, -CD59, and -CD16 was performed on all patients and controls to assess the prevalence of deficiencies and PNH-type phenotype clone size. Deficiencies of individual GPI-AP were seen in 17.3% cases versus 3.4% controls. This was due to CD55 deficiency on red blood cells and CD16 deficiency on the granulocytes. Deficiency of multiple GPI-APs was less frequent (3.7% cases). Data of this study indicate that the PNH defect as detected with CD55, CD59, and CD16 is not an important cause of intra-abdominal thrombosis in northwestern India.
Introduction
Thrombotic disorders are a common cause for morbidity and mortality in adults. A variety of hereditary and acquired factors have been implicated in the causation of venous thrombosis. Among the better known hereditary factors are the factor V Leiden mutation, protein C, protein S, and antithrombin III deficiency. Acquired factors include myeloproliferative disorders, anticardiolipin antibodies (ACA), and lupus anticoagulant. Paroxysmal nocturnal hemoglobinuria (PNH) is a rare hematological condition that has a high incidence of thrombosis. It is characterized by intravascular hemolysis, cytopenias with marrow aplasia and venous thrombosis. 1 Nearly one fourth of patients with PNH in the Western world have thrombosis and these account for 50% of all deaths in patients with PNH. Intra-abdominal veins are the most common sites of thrombosis in patients with PNH. Hepatic venous thrombosis (Budd Chiari syndrome [BCS]) is a serious, potentially fatal complication of PNH. 2 –5 In various series, 15% to 40% of patients with PNH had hepatic venous obstruction. 1,3,4,6 –8 However, literature on PNH being diagnosed in primary thrombotic presentation is scarce. Most current data stem from retrospective series and case reports. 9 –12 The largest prospective study, till date, comprises 163 patients with BCS from 9 different European countries (EN-Vie study cohort). 13 In this study, PNH was detected in approximately 19% of cases of BCS. It has been emphasized in the recent guidelines 14,15 that testing for PNH in patients with BCS needs to be part of the standard etiology screening, as diagnostic signs may not be very specific, for example, only slight anemia.
Since all patients with PNH may not have hemolysis, this diagnosis may be missed unless specifically looked for. Intra-abdominal thrombosis, being a preferred site of venous thrombosis in PNH, therefore, presents an opportunity to detect this disease. The diagnosis of PNH as an underlying cause for thrombosis has clinical implications. Patients with venous thrombosis, including some with BCS receive warfarin as part of therapy. The anticoagulants may be discontinued, if no other factor predisposing to thrombosis is found. 16,17 In chronic disorders like PNH, this may be potentially harmful and predispose the patient to recurrent thrombotic events. The curative treatment of PNH entails marrow transplantation; hence, it is important to ascertain whether PNH is an etiological factor in a patient with thrombosis. We thus attempt to answer the question whether PNH defect is prevalent in patients with thrombosis in the intra-abdominal vessels, that is, hepatic veins (BCS) and extrahepatic venous obstruction (EHPVO).
Paroxysmal nocturnal hemoglobinuria is a clonal stem cell disorder characterized by deficiency of glycosylphosphatidyl inositol-anchored proteins (GPI-APs). 18 The GPI-APs include complement regulatory proteins like decay accelerating factor (CD55), membrane inhibitor of reactive lysis (CD59), and other membrane proteins participating in immune functions (CD14, CD16, CD66b, and CD67). Flowcytometric analysis using antibodies directed against GPI-APs is now the most sensitive and informative assay available for the diagnosis of PNH. 14,15,19 –21 Quantification of atleast 2 GPI-APs is recommended to exclude the possibility of a rare inherited, isolated deficiency of a single GPI-AP. 22 CD55, CD59, and CD16 are reliable markers for flowcytometry. 15 It is well established that thrombosis has a multifactorial etiology and a combination of prothrombotic factors has a synergistic effect on the causation of thrombosis. The thrombophilic states have varied distribution in ethnically different populations. The prothrombin gene mutation G20210A is virtually nonexistent in the Indian subcontinent. 23 Testing for the thrombophilic states incurs a high cost and this is a major issue for patients in resource constraint settings. Paroxysmal nocturnal hemoglobinuria being a rare disorder, there is presently no clear opinion on inclusion of flowcytometry in the screen for thrombophilic defects. We screened patients with intra-abdominal thrombosis for PNH phenotype using flowcytometry on granulocytes and RBCs to assess the presence of PNH defect in this population and consider the inclusion of this test in the routine thrombophilia screen.
Materials and Methods
This was a prospective cross-sectional study. Investigations were performed in the Department of Hematology, Postgraduate Institute of Medical Research and Education, Chandigarh (PGIMER), India, on patients registered with the Liver Clinic of department of Hepatology, Nehru hospital, PGIMER, Chandigarh. Forty-four patients with BCS and 37 patients with EHPVO were enrolled during a period from October 2008 to October 2011. Patients with obstruction to hepatic veins at any site from efferent vein of lobules to the entry of inferior vena cava into right atrium, with the clinical presentation of hepatomegaly, abdominal pain, and ascitis were included in the category of BCS. 24 Doppler ultrasonography showing hepatic vein abnormalities such as lack of visualization of normal hepatic venous connections to inferior vena cava, comma-shaped intrahepatic or subcapsular collateral vessels, and absence of flow signal in the hepatic vein were included. Cases with acute presentation of BCS were excluded.
Extrahepatic portal vein obstruction was confirmed with abdominal imaging that showed echogenic thrombus in the portal vein, extensive collateral vessels in the porta hepatis, an enlarged spleen and nonvisualization of the portal vein. In cases with inconclusive ultrasonography, the same was confirmed by computed tomography or magnetic resonance imaging studies. The concomitant clinical picture was of previous history of upper gastrointestinal bleeding, abdominal pain, or distention. Totally, 87 healthy controls were also included. Two milliliters of venous blood samples from patients and controls were collected in tubes containing EDTA. All samples were analyzed within 24 hours of collection. Prior consent was obtained from the patient and samples were drawn as far as possible on the day of bleeding for prothrombotic factors. The same sample was subjected to flow cytometry with fluorochrome-conjugated monoclonal antibodies on RBCs and granulocytes. Laboratory data regarding the prothrombotic work-up in patients were obtained. The study was approved by the Institute Ethics Committee.
Flowcytometry
Granulocytes
To detect the expression of GPI-AP on granulocytes the following monoclonal antibody reagents were used: CD55 conjugated with phycoerythrin ([PE] Cat No. 555574, BD Pharmingen Clone IA10), CD59 conjugated with fluorescein isothiocyanate (Cat No. 555573, BD Pharmingen Clone p282 H 19) and CD16 conjugated with PE (BD 347617 clone B73.1).
A lyse–wash–stain technique was followed. Two milliliters of whole blood aliquots were taken. The RBCs were lysed by addition of ammonium chloride-based lysing agents. Following incubation for 10 minutes at room temperature, it was centrifuged for 5 minutes at 660g. The supernatant was removed and the cell pellet was washed twice using 2 mL of phosphate-buffered saline (PBS). The pellet was resuspended in 500 µL of PBS. Then a 50 µL aliquot of this sample was incubated with 10 µL of fluorochrome-conjugated monoclonal antibodies for 30 minutes at room temperature in dark. The antibodies used had been standardized in terms of amount and procedure on known cases with PNH presenting with cytopenias or hemolysis.
Red blood cells
The expression of CD55 and CD59 in RBCs was studied in 20 µL of whole blood. Ten microlitres of fluorochrome-conjugated monoclonal antibodies were added and incubated at room temperature in dark for 30 minutes. After washing with PBS, the supernatant was discarded. The cell button was resuspended in 500 µL of PBS. CD16 is not present on RBCs and hence was excluded from the panel used in RBCs.
Acquisition and Analysis
The analysis was performed on BD FACS Calibur flowcytometer with Cell Quest software, taking atleast 10 000 events (cells). Granulocytes were gated using their forward scatter/side scatter characteristics. For RBCs, a gating strategy based on forward scatter and side scatter amplification in log mode was applied. Both dot-plots and histograms of cell counts versus log fluorescence were obtained.
Appropriate normal and negative controls (an isotype-matched monoclonal antibody) were run in parallel to the patient’s sample. The proportions of positive and negative cells were recorded. The presence of >10% of cells deficient in GPI-linked proteins was taken to indicate PNH defect.
Thrombophilia Workup
Patients who were not on oral anticoagulants (37 cases of BCS and 27 cases of EHPVO) underwent screening for thrombophilic factors. These included the functional assays of protein C (STA Clot Protein C, Diagnostica Stago), protein S (STA Clot Protein S), chromogenic assay for antithrombin III (STAchrom Antithrombin, Diagnostica Stago), and lupus anticoagulant (LA, DRVVT based Screen and Confirm, Siemens). All these were performed on the STA Compact Coagulation analyser (Diagnostica Stago, France). Screening for ACAs was done by enzyme-linked immunosorbent assay (Orgentec, GmBh). The DNA-based testing for factor V Leiden mutation (FVL) was done on peripheral blood lymphocytes.
Results
Overall, 81 patients (44 with BCS and 37 with EHPVO) and 87 control subjects were screened for PNH by flow cytometry with CD55, CD59, and CD16.
None of them had history of hemolysis, jaundice, recent red cell transfusions, or raised unconjugated bilirubin levels at the time of testing. There was no history of passing cola-colored urine in the past. Anemia was present in 82%, leukopenia in 23%, and thrombocytopenia in 34% of all cases. None of the controls had anemia, leukopenia, or thrombocytopenia.
Flow cytometry
Fourteen (17.3%) cases had at least one deficient GPI-AP on testing versus 3 (3.4%) controls. Majority of these had a single GPI-AP deficiency on either the granulocytes or RBCs. Presence of more than 1 deficient GPI-AP on same cell/or single GPI-AP deficient on 2 cell types was seen in 3 (3.7%) cases. However, none of the controls showed the presence of any combination of markers deficient on granulocytes or RBCs (Table 1).
Distribution of Patients and Controls With More Than 1 Marker Deficient on RBCs or Granulocytes in the Study Group.

Abbreviations: BCS, Budd Chiari syndrome; EHPVO, extrahepatic venous obstruction; GPI-AP, glycosylphosphatidyl inositol-anchored protein; RBC, red blood cell.
CD 55 deficiency on the RBCs and CD 16 deficiency on granulocytes was seen in cases of EHPVO, BCS as well as controls. On the RBCs, the CD55 staining was of lower intensity when compared with CD59. It was not possible to get good separation between the truly negative cells and dim positivity of CD55 on RBCs. The mean CD55 deficient clone size in RBCs was 1.9% (range 0-8.8%). Using the standard cutoff of >1% deficient cells as positive for PNH phenotype was associated with an unacceptably high number of positives in control participants (47 of 81, 58%) The best discrimination between cases and controls was seen using >10% deficient cells as the cutoff. The CD59 staining on RBCs was uniformly bright. The number of cases with CD55-deficient RBCs and the mean clone sizes in these deficient samples were comparable in all the patient groups. The widest range of deficiency was seen with CD16 on the granulocytes—ranging from 10.83% to 86.9% cells with deficient phenotype. A single case of BCS having a CD59-deficient phenotype on RBCs was present in the patient cohort. The CD59-deficient phenotypes were not seen in cases of EHPVO and controls (Table 2).
Flowcytometry Parameters Showing the Deficiency of the GPI-AP–CD55, -CD59, and -CD16 on RBCs and Granulocytes and the Mean and Maximum Size of Deficient Phenotypes.a
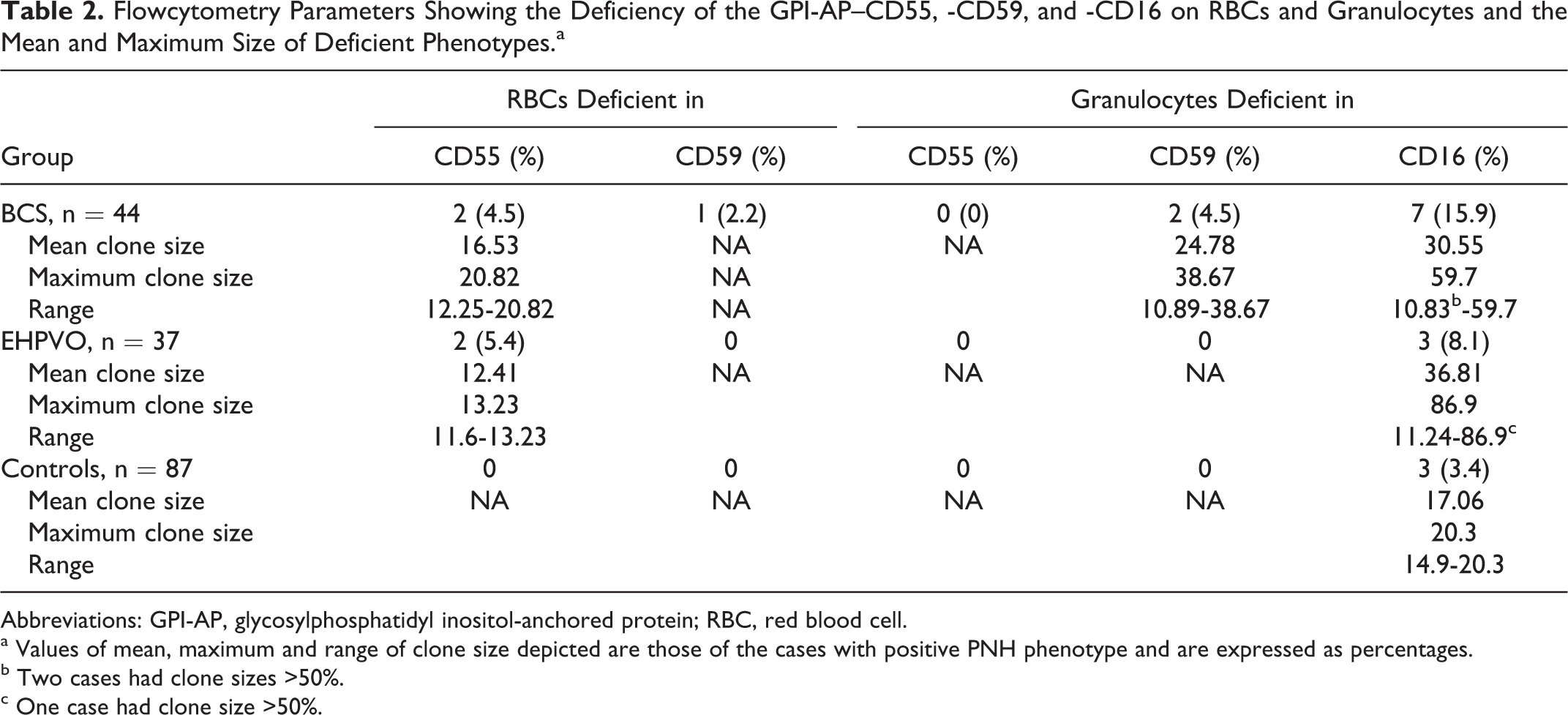
Abbreviations: GPI-AP, glycosylphosphatidyl inositol-anchored protein; RBC, red blood cell.
a Values of mean, maximum and range of clone size depicted are those of the cases with positive PNH phenotype and are expressed as percentages.
b Two cases had clone sizes >50%.
c One case had clone size >50%.
In the entire study group, only 3 (2 cases with BCS and 1 case with EHPVO) of 81 patients had atleast 2 markers deficient on more than 10% cells (Table 3). However, none of these patients fulfilled the diagnostic criteria of PNH, in terms of deficiency of atleast 2 different monoclonal antibodies, directed against 2 different GPI-APs, on 2 different lineages.
Combinations of Deficient Phenotypes on RBCs and Granulocytes.
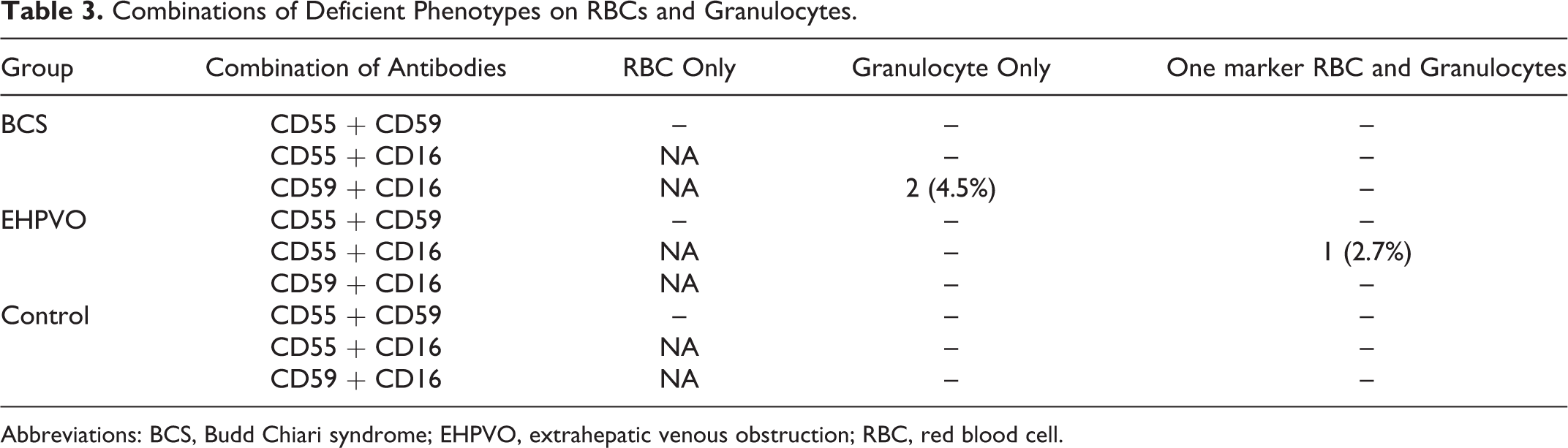
Abbreviations: BCS, Budd Chiari syndrome; EHPVO, extrahepatic venous obstruction; RBC, red blood cell.
Thrombophilia Parameters
The thrombophilia factors were tested in 37 (84%) cases of BCS and 26 (72%) of EHPVO. In the remaining cases, the prothrombotic workup was deferred due to ongoing oral anticoagulant therapy or recent transfusion with plasma or could not be completed since some patients were lost to follow-up. Protein S deficiency (41.7% and 33.3% cases of BCS and EHPVO each) was the commonest followed by protein C deficiency (33% and 11.1% cases of BCS and EHPVO each). Antithrombin III deficiency was seen in 17.1% cases of BCS and 19.2% cases of EHPVO. Anticardiolipin antibodies and FVL heterozygosity were rare. None of the cases showed positivity for lupus anticoagulant (Table 4).
Profile of Thrombophilic Factors in Patients With BCS and EHPVO.

Abbreviations: BCS, Budd Chiari syndrome; EHPVO, extrahepatic venous obstruction.
Flow cytometry screening of patients with non thrombotic presentation for suspected PNH
During the study period, 255 cases were screened for suspected PNH with nonthrombotic presentation. These included cases with cytopenias, myelodysplastic syndromes, and hemolytic anemia. The methodology of testing, reagents, and cutoff criteria for deficient cells was kept same. Fifteen (5.5%) of these fulfilled the criteria for PNH (Table 5).
PNH-Type Phenotype on Flowcytometry in Nonthrombotic Presentations.a
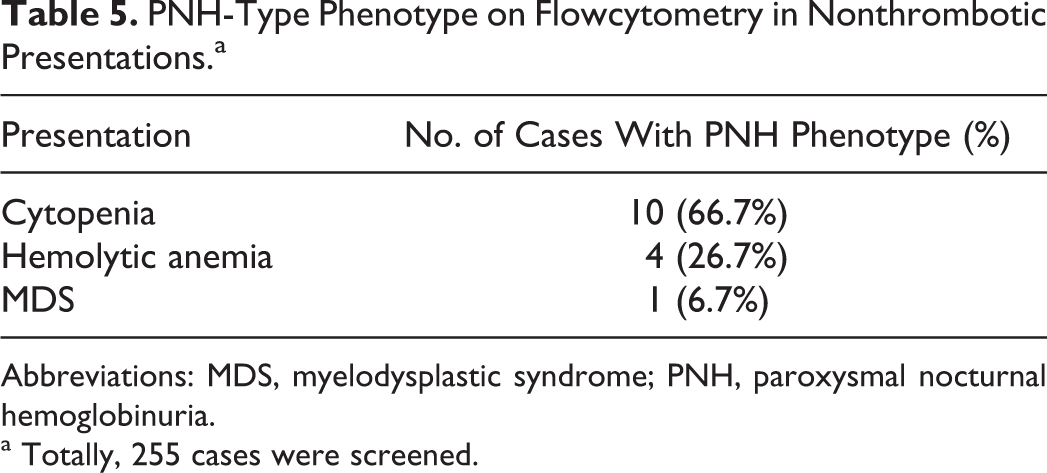
Abbreviations: MDS, myelodysplastic syndrome; PNH, paroxysmal nocturnal hemoglobinuria.
a Totally, 255 cases were screened.
Discussion
Most modern laboratories are able to provide a fairly extensive workup for thrombosis. Despite technical advancement and refinement in the molecular diagnostic techniques, no cause can be found in nearly 30% to 40% of patients with venous thrombosis. Hence, there is an ever expanding list of tests that can be performed for thrombosis. Some uncommon disorders like PNH and myeloproliferative neoplasms have been associated with thrombosis. Paroxysmal nocturnal hemoglobinuria is an unusual disorder, in that it has a peculiar effect on the coagulation system. On one hand, it is associated with cytopenias especially thrombocytopenia and should favor bleeding, whereas on the other hand, thrombosis is a well-reported complication of PNH. In addition, PNH tends to produce thrombosis at unusual sites with a predilection for the intra-abdominal and intracranial venous vasculature. Being a rare disorder, testing for PNH get overlooked in the workup of venous thrombosis. There are a number of reports on the role of thrombophilic factors in 2 intra-abdominal thrombotic disorders—EHPVO and BCS, however, the literature on PNH being diagnosed in a primarily thrombotic presentation is scarce. It is our hypothesis that PNH may be an important cause of intra-abdominal thrombosis. Interestingly, EHPVO is often accompanied by cytopenia and for practical purposes it fulfils 2 screening criteria (thrombosis and cytopenia) commonly used in PNH screening.
We studied the prevalence of PNH phenotype by assessing the expression of CD55, CD59, and CD16 on the RBCs and granulocytes in the patients with intra-abdominal thrombosis.
The analysis of GPI-linked antigen expression on erythrocytes and peripheral blood granulocytes by flow cytometry has become firmly established as the technique of choice for screening and diagnosis of PNH, replacing many of the original biochemical assays that relied upon demonstrating the in vitro susceptibility of PNH red cells to complement-mediated hemolysis. It is now considered the gold standard in diagnosis of PNH. A comparison of variety of methods has outlined the usefulness of these techniques. 25 Conventional screening assays are time consuming and need standardization.
In this study, a minority of cases had at least one deficient GPI-AP marker on the cells in the patient group (14 cases, 17.3% vs 3.4% controls). Two of the GPI-AP antigens, that is, CD55 on RBC and CD16, on the granulocytes contributed the bulk of the deficient phenotypes seen. Virtually, all the commonly used antibodies for PNH testing have their specific strengths and weakness attributable to the type of clone used, at what stage of maturation the antigen is expressed and its strength of expression. 26 Of these GPI-APs, the CD55 and CD59 complement activation regulatory proteins are commonly used in the flowcytometry-based diagnostic screening for PNH. This is related to the fact that (1) both proteins are expressed universally in the different cell populations present in normal peripheral blood and (2) abnormal expression of CD55 and CD59 correlates with the clinical behavior of the disease. 21,27 Previous reports have shown that CD55 expression on the RBCs is lower than that of CD59 and this often leads to problems in assessing the PNH-type phenotype using CD55 on RBCs. 5,22 In our cases though frequently seen, CD55-negative RBCs failed to form discrete populations either on dot-plot scattergrams or histograms. The range of RBCs with the PNH-type phenotype extended from 11.6% to 20.8%. However, in the case with the highest PNH-type RBC fraction using this antibody, we were unable to locate a discrete peak well separated from the normal population. Recent guidelines 15 recommend a cutoff of >1% GPI-AP-deficient cells to be labeled as a PNH clone. In this study, using this cutoff, we found an unacceptably high number of PNH phenotype-positive controls especially for CD55 on RBCs and CD16 on granulocytes. Since the gating strategy did not utilize a non GPI-linked anchoring marker for identifying neutrophils, that is, CD15 and CD235 for RBCs, we used a higher cutoff, that is, >10% deficient cells to assign the PNH-positive phenotype. Admittedly this approach might have decreased the number of cases identified by positive clones, however, it ensured that no normal subjects were wrongly labeled positive for PNH clone. Recent guidelines do not recommend screening for CD55 on RBCs, possibly as a result of the inherent low intensity staining on the RBCs. 15 In the present study, in contrast, CD59 staining on the RBCs was bright and uniform. In a single case of BCS with a small PNH-type population with CD59-deficient RBC in the entire case cohort, this finding was not duplicated on the patient’s granulocytes or in other GPI-APs that we tested.
Hernandez et al 26 observed that the reactivity for CD55 on most subsets of leukocytes, except the eosinophils, was higher or similar to that of CD59, pointing out the potential greater utility of the former antigen for the identification of PNH cells in most populations of leukocytes. In the present study, CD55 staining on the granulocytes was bright conforming to what has been reported previously. Various studies have reported a strong correlation between a larger PNH clone and occurrence of thrombosis. 1,4 Hall et al 4 showed that the 10-year risk of thrombosis in patients with more than 50% GPI-AP-deficient granulocytes was 44% compared with 5.8% for patients with less than 50%. In a study by Moyo et al, 1 14 patients (29%) of the total of 49 patients studied developed thrombosis. They reported a strong association between the size of the PN clone (as measured in granulocytes) and thrombosis. No patient with <61% PNH granulocytes developed thrombosis, whereas 12 of 22 patients (54.5%) with ≥61% PNH granulocytes developed a thrombosis. Using a logistic regression model, for a 10% change in PNH clone size, the odds ratio for risk of thrombosis was estimated to be 1.64. In the entire study group, only 3 (2 with BCS and 1 with EHPVO) of 81 patients had at least 2 markers deficient on more than 10% cells. However, none of these patients fulfilled the diagnostic criteria for PNH (deficiency of atleast 2 different monoclonal antibodies, directed against 2 different GPI-APs, on 2 different lineages). Both cases of BCS had CD59 and CD16 deficient PNH-type phenotype on the granulocytes; however, there was no deficiency of CD59 on the RBCs in either case. There was no history of a red cell transfusion and no laboratory evidence of recent hemolysis to explain this discrepancy. The CD55 deficiency was not seen in the RBCs or granulocytes of these cases. A single case of EHPVO had 2 different GPI-AP deficiencies, that is, CD55 on the RBCs and CD16 on the granulocytes. Since CD55-deficient phenotype was absent on the granulocytes, it was difficult to label this case as PNH with surety. The relatively small size of the deficient phenotype 13.23% (CD55 on the RBCs) and 12.31% (CD16 on the granulocytes) also does not support the known fact about thrombosis in PNH occurring with large granulocyte clone sizes. Paroxysmal nocturnal hemoglobinuria-type phenotype with CD55 staining on granulocytes was not seen in patients or controls.
Antibodies such as CD16, CD24, and CD66b work equally well for granulocyte analysis. A combination of CD16/CD66b has the added advantage of clearly separating granulocytes from eosinophils and is shown to be superior to CD55/CD59 in screening subclinical PNH where the number of neutrophils may be less. 28 However, this study was aimed at looking at clinical PNH presenting as thrombosis, where the clone sizes are expected to be large. Hence, this combination was not considered mandatory. Both CD16 and CD66b are expressed late in myeloid maturation, and their detection enables a clear resolution between PNH cells (negative populations) and normal granulocytes (positive populations). This combination is useful for examination of bone marrow. In the present study, the PNH-type phenotype with CD16 staining of granulocytes was seen in 12.3% cases. Three of these had large fractions (>50%). These cases were not associated with PNH phenotype by the other markers and hence, do not meet the criteria of PNH. It has been noted that in patients who had isolated granulocyte PNH phenotypes, massive hemolysis of affected RBCs may lead to failure in detecting deficient clones on RBCs on flowcytometry. Alternatively, a recent transfusion with normal RBCs may mask the underlying PNH clone. Both of these factors were excluded in these cases on the basis of history and lack of laboratory evidence of hemolysis. The CD16 deficiency was also seen in 3.4% of controls. The disadvantage of using CD16 and not CD16b is that CD16 by itself is expressed in nongranulocytic series of leukocytes (natural killer [NK] cells and macrophages) and therefore, there is need for an additional marker for sensitive gating. However, as the population under study was not anticipated to have aplastic marrows, the number of granulocytes available for analysis was expected to be adequate and CD16 was included in the antibody panel for granulocytes. Samples with proportionately high eosinophils have a large CD16-negative component as this antigen is not constitutively expressed on eosinophils. Peripheral smear examination revealed increased eosinophils in 2 cases of BCS and 1 of EHPVO and may have contributed to the increased CD16 deficiency seen in the events gated. Richards et al 20 highlighted that CD16 if used in isolation can cause confusion and potential misdiagnosis. An infrequent polymorphism in the CD16 determinant renders the antigen undetectable to some monoclonal antibodies and not others. The presence of a genetic polymorphism was not confirmed in this study.
The CD16 clone B73.1 (BD 347617) used in the present study reacts with neutrophils at a lower intensity than CD16 clone NKP 5 and CD16 clone G022. However, majority of the cases in the present study did show bright CD16 on granulocytes.
FLAER is universally recognized to have great value in the detection of PNH clones in granulocytes and monocytes; however, it is still not easily available in all parts of the world. FLAER staining was used in a number of subsequent cases (3 cases with BCS and 7 cases with EHPVO), not included in this study, and the largest PNH-type population seen with was of the size of 1.4%. The small sample size with FLAER staining prevents us from drawing definite conclusions; however, a trend toward predominantly negative cells is anticipated and needs to be confirmed on a larger group.
Alongside with this study, we assessed the occurrence of PNH phenotype in patients referred for flow cytometry for nonthrombotic presentation of PNH, that is, with pancytopenic and hemolytic features. The methodology for flowcytometry remained the same. Two hundred and fifty-five cases were screened for PNH by flowcytometry in the study period. Of these, 15 (5.8%) patients were found to have PNH. None of these patients had thrombosis at or prior to presentation. This is in contrast to the western literature where about 40% of the PNH have thrombosis. The most feared complication of PNH, venous thrombosis, occurs in 50% of patients with hemolytic disease and is the cause of death in at least one third of such patients. 2,5 There is a predilection for the intra-abdominal and cerebral veins. The risk of thrombosis is significantly higher in patients from Europe and the United States than in patients from the Far East. 14 There also appears to be an increased risk of thrombosis in African American or Latin American patients. 29 A possible explanation is that patients from different ethnic groups may have different additional inherited prothrombotic traits; however, no correlation between the inherited thrombophilia and thrombosis in PNH could be demonstrated.
In this study, proteins C and S deficiency were the most common thrombophilic defects seen. The data are in concordance with that reported in literature 23,30 with some differences. Factor V Leiden mutation is the commonest inherited thrombophilia defect in the West (10%-37% in venous thrombosis patients and 0.45%-10% in general population). However, in Asia, its prevalence is low. The prothrombin G20210A mutation is present in 0.7% to 4% of the general population and up to 8% of venous thrombosis patients in Western countries, while in India and many other Asian countries, it has not been detected and screening for this mutation is generally not indicated in these populations. With regard to the natural inhibitors of coagulation (protein C, protein S, and antithrombin), there is a wide range in the prevalence of their deficiencies partly because of a secondary hepatic insufficiency which develops in these patients. PC was the only significantly associated factor with both PVT and BCS, respectively, in one study, whereas another did not show a significant difference in the prevalence of PC, PS, and AT when compared with the normal controls, DVT, and EHPVO cases. 31 Although the prevalence of PS and AT deficiency is low in some series of BCS and PVT, 32,33 their role in the pathogenesis of BCS and PVT is difficult to exclude and there are several case reports to this effect. 34,35 Antiphospholipid antibodies have been detected in 10% of patients in India.
There is limited literature on PNH in India. Intra-abdominal thrombosis was reported in 2 and 1 cases in cohorts of 104 and 11 cases patients of PNH, respectively. 12,36 None of the 17 cases of PNH had venous thrombosis in another report from North India. 37 In the study addressing the clinical profile of the children with PNH diagnosed over 6 years, only a single female presented with arterial thrombosis. 38 Gupta et al 39 in a series of 18 patients with hepatic vein obstruction found that 3 had PNH.
It is possible that PNH in this region does not have a prominent thrombotic presentation; however, it needs to be borne in mind that some of the Indian series have reported PNH based on diagnosis by methods other than flowcytometry. A sensitive technique like flowcytometry may be able to detect more cases with an additional advantage of granulocytic analysis for PNH defect.
The limitation of the present study includes the modest sample size in the patient group. Present guidelines have recommended the use of FLAER and CD55 on granulocytes. The latter has been used in this study. A pilot sample tested with the former shows a trend toward negativity for PNH-type phenotypes. Red blood cell staining with CD55, although performed in this study, is no longer recommended. The data of this study would support this guideline, since CD55 results were difficult to interpret and showed nonspecific PNH-type phenotypes in control samples as well. In fact the RBC screening for PNH by flowcytometry is now not recommended upfront, but to be performed if a clone is detected on granulocytes.
The data of this study indicate that PNH defect as detected by flowcytometry with CD55, CD59, and CD16 is not an important cause of intra-abdominal thrombosis leading to BCS or EHPVO, either in isolation or in combination with other thrombophilic factors. Thrombotic manifestations of PNH are associated with large granulocyte clone sizes, which were not seen in this study. However, it would be prudent to examine a larger population with intra-abdominal thrombosis before discarding the policy of screening such patients for PNH. Further, data with clone sizes measured by FLAER-based flowcytometry would help to confirm these results. In a resource constraint setting, flowcytometry for PNH may be a low priority screening for a thrombophilic state in patients with intra-abdominal thrombosis.
Footnotes
Acknowledgment
The technical assistance provided by Ms Praveen Bose in the performance of flowcytometry is gratefully acknowledged.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.