
Abstract
Acute painful crisis is a common sequela that can cause significant morbidity and negatively impact the quality of life of patients with sickle cell disease (SCD). Plasma levels of several chemokines and cytokines including tumor necrosis factor-α (TNF-α), interleukin 1β (IL-1β), IL-6, IL-8, monocyte chemoattractant protein 1 (MCP-1), macrophage inflammatory protein 1α (MIP-1α), and interferon γ (IFN-γ) in patients with SCD showed a distinct and statistically significant rise either during painful crisis or at steady state. Plasma levels of various growth factors, including human vascular endothelial growth factor (VEGF), human basic fibroblast growth factor (FGF), and human hepatocyte growth factor (HGF), showed a sustained 2- to 3-fold increase either during painful crisis or at steady state in patients with SCD. Furthermore, plasma levels of the biomarker
Keywords
Introduction
The clinical hallmark of sickle cell disease (SCD) is the painful acute “crisis” that has, in spite of therapeutic advances, continued to be a treatment challenge. Such crises occur with variable frequency and duration, and they commonly require hospitalization. 1 The pathobiology of SCD is instigated by episodic vascular occlusion, with multiple inciting events including hemoglobin S (HbS) polymerization and mechanical obstruction induced by sickled red blood cells (RBCs). An important pain–induction mechanism is thought to involve bone marrow vasculature infarction, leading to the release of certain inflammatory mediators, which in turn stimulate afferent nerve fibers and cause pain. Vaso-occlusion also involves adherence of circulating blood elements such as leukocytes to endothelial cells, hypercoagulability, endothelial dysfunction,2,3 and altered nitric oxide (NO) metabolism and ischemia–reperfusion injury. 4
Abnormal interactions between erythrocytes and endothelium are a primary initiating factor in the development of microvascular occlusions in SCD. This view is supported by the significant correlation between clinical severity of SCD and extents of RBC adhesiveness.2,5 Neutrophils are likely to be an important factor in causing microvascular sickle cell trapping and consequent vaso-occlusion; sickle cells appear to be more adherent to neutrophils than to normal red cells. Sickle cells also increase neutrophil oxidative activity, which may be important in neutrophil-induced tissue damage during vaso-occlusive episodes. 6
Patients with SCD exhibit high plasma levels of markers of thrombin generation, depletion of natural anticoagulant proteins, abnormal activation of the fibrinolytic system, and increased tissue factor expression, even in the noncrisis steady state.
7
In addition, platelets and other cellular elements are chronically activated, which might predispose the patient to thromboembolic manifestations.8–10 Sickle cell disease is often referred to as a hypercoagulable state because patients manifest increased thrombin and fibrin generation and increased tissue factor procoagulant activity.11,12 Finally, increased levels of measured plasma markers of coagulation activation (
A central aspect of sickle cell vasculopathy is the impairment of endothelial regulation of vasomotor tone, thrombosis, and inflammation. 2 Occurrence of intermittent vascular occlusion in SCD leads to reperfusion injury associated with granulocyte accumulation and enhanced production of reactive oxygen species. The participation of NO in oxidative reactions causes a reduction in NO bioavailability and contributes to vascular dysfunction in SCD. 3
There is emerging consensus that a proinflammatory condition contributes to the vaso-occlusive complications of SCD. Tissue damage due to vaso-occlusion releases numerous inflammatory mediators that initiate the transmission of painful stimuli and the perception of pain. Cytokines derived from platelets, white blood cells, and endothelial cells have all been implicated in the development of several sequelae of this disease.14,15 Thus, in as much as sickle cells’ adhesion to the endothelium plays a role in sickle cell vaso-occlusion, the presence of such diverse mechanisms of adhesion involving selectin, integrin, and immunoglobulin present an enormous challenge for delineating physiologically relevant therapeutic targets. Interestingly, recent studies have suggested that targeting a specific adhesion pathway such as selectins may be sufficient to reduce vaso-occlusion. 16 The present investigation examined the role of various proinflammatory cytokines, chemokines, growth factors, and coagulation parameters in patients with SCD during active crisis at admission and prior to the administration of analgesic and at steady state as compared to age-matched, healthy volunteers.
Methods
Clinical Profile of Enrolled Patients With SCD
The participants of this study were admitted through the emergency room at King Abdel-Aziz Hospital, with an established diagnosis of painful vaso-occlusive crisis of sickle cell anemia (SCA). Sickle cell anemia was proved by alkaline electrophoresis of hemoglobin. 17 All participants were adults over the age of 18, divided into 3 groups, and all signed a consent form in accordance with an internal review board–approved protocol at King Abdel-Aziz Hospital, Jeddah, Saudi Arabia. Participants enrolled in the study include painful crisis group comprising 36 patients, steady state group comprising 30 patients, and the control group comprising 35 healthy, age- and sex-matched donor patients (Table 1 ). All patients enrolled in this study were explained on the nature of the study and were consented.
Healthy and Sickle Cell Human Participants Enrolled in the Study
a NS, Not statistically significantly different from healthy participants and between groups (a) and (b).
The inclusion criteria were the proven diagnosis of SCA confirmed by alkaline hemoglobin electrophoresis, and the painful crisis was assessed by a hematologist to rule out the presence of other causes of pain that can mimic painful crisis of SCA or any medication apart from folic acid used on a long-term basis in patients with SCA. The sampling of blood took place at the height of pain in the emergency room prior to admission and before the institution of any analgesic or any other medication. The participants of the steady state group were proven to have SCA in the same manner as before, and they were found to have no painful crisis in the past 30 days or any pain at the sampling time. Exclusion criteria were that the patients who were on any medications for any other coexisting condition were excluded from the study. Patients who were pregnant or having other conditions at the sampling time were also excluded. Patients who were found to have any other coexisting disease during their course of hospitalization had their samples removed from the study.
Blood drawing was carried out using a butterfly needle and the first 2 mL were discarded, and then 10 mL blood samples were collected in 3.2% citrate tubes. Basal blood samples were collected (10 mL in citrate tubes, 3.2%) from patients with SCD during acute painful crisis and at steady state when the patients were in stable condition. Additionally, blood samples were collected from healthy age-matched volunteers in the same manner as above. Blood samples were centrifuged at 5000g for 15 minutes, and plasma samples were transferred into coded tubes and frozen at −60°C until assayed for the different biomarkers. Different biomarkers were measured using bioplex (Bio-Rad Laboratories, Inc, Hercules, California); tumor necrosis factor-α (TNF-α) and
Statistical Analysis
Analysis of variance was conducted using the Kruskal–Wallis Test. Proportions from 2 or more independent groups were compared using either the chi-square test or the Fisher Exact test, as appropriate. A P value ≤.05 was considered statistically significant.
Results
Plasma TNF-α levels were significantly elevated in patients either during painful crisis or at steady state, as compared to levels in patients of the healthy, age-matched control group (P < .01). Surprisingly, TNF-α levels were significantly higher at steady state (P < .05), as compared to levels during painful crisis (Figure 1A). Plasma interleukin 1β (IL-1β) followed the same trend as that of TNF-α, with significant elevation of levels during painful crisis as compared to levels in patients of the healthy, age-matched control group (P < .01), and a much higher level at steady state (P < .05) as compared to levels during painful crisis (Figure 1B). Plasma IL-1β levels were elevated by 100- to 200-fold above levels in patients of the healthy, age-matched control group (Figure 1B). Similarly, plasma IL-6 levels followed the same trend as TNF-α and IL-1β levels, with a 20- to 70-fold rise above that of patients in the healthy, age-matched control group (Figure 1C). Additionally, IL-8 plasma levels in patients with SCD showed a distinct rise (P < .01) that did not change between patients with SCD, either during painful crisis or at steady state, with a 30- to 50-fold increase above that of healthy, age-matched control group (Figure 1D). These data suggest an upregulation of inflammatory cytokine and chemokine production at a common transcription pathway, perhaps at the nuclear factor κB (NF-κB) junction.
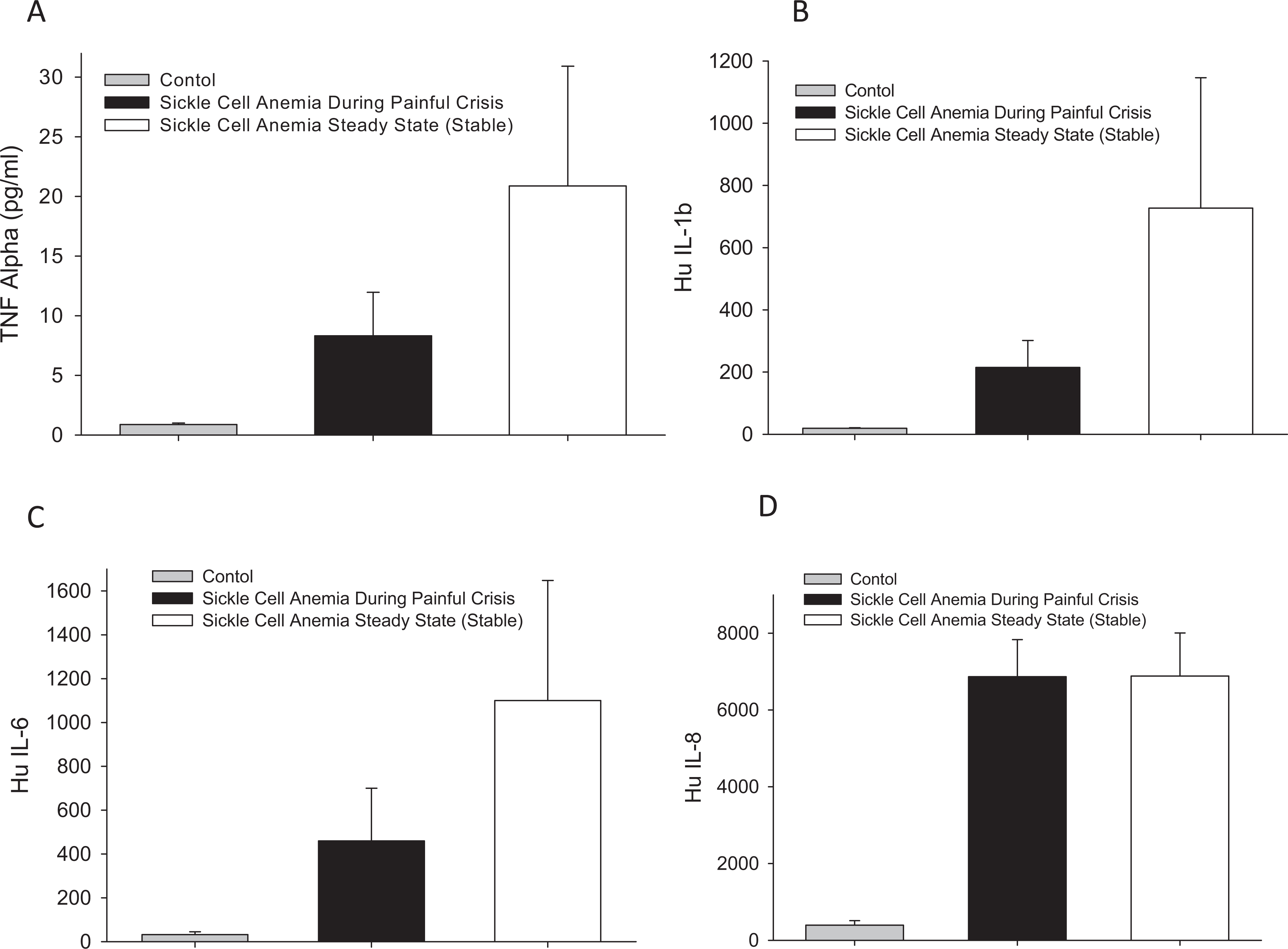
Plasma levels of different inflammatory mediators in patients with sickle cell disease during crisis and at steady state, as compared to age-matched, healthy participants (A) TNF-α, (B) IL-1β, (C) IL-6, and (D) IL-8. TNF-α indicates tumor necrosis factor-α; IL, interleukin.
Plasma levels of monocyte chemoattractant protein 1 (MCP-1) in patients with SCD showed a distinct rise (P < .01) either during painful crisis or at steady state, with a 20- to 30-fold increase (Figure 2A) and macrophage inflammatory protein 1α (MIP-1α) increased by about 3-fold above that of healthy, age-matched control group (Figure 2B). In contrast, eotaxin plasma levels did not show statistically significant changes between patients with SCD, either during painful crisis or at steady state compared to the levels in the healthy, age-matched control group (Figure 2C). However, IL-10 plasma levels were modestly elevated by 1.5- to 2-fold in patients with SCD either during painful crisis or at steady state (P < .05), as compared to that of healthy, age-matched control group (Figure 2D). These data suggest differential upregulation of cytokines and chemokines to different degree in patients with SCD.
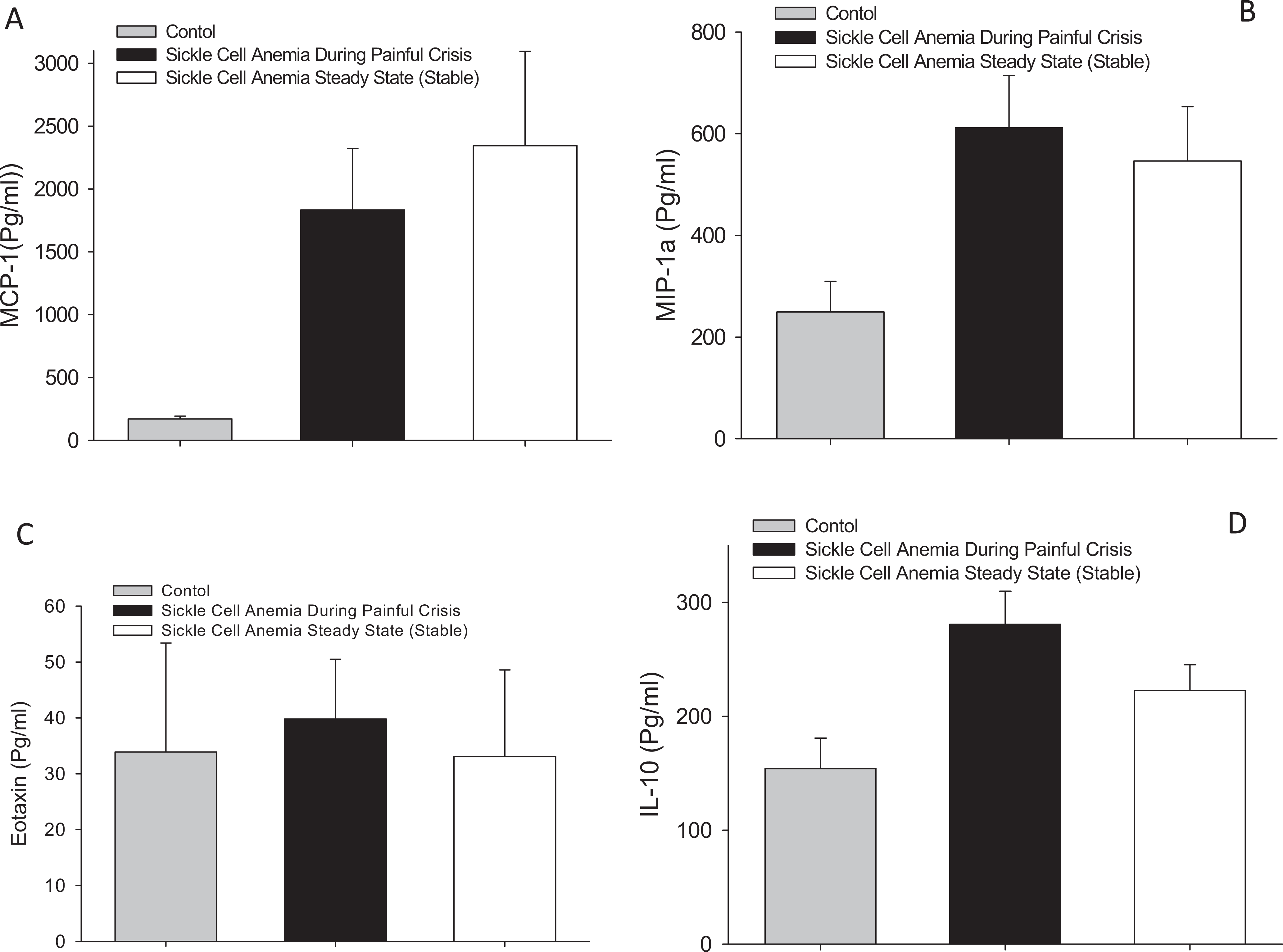
Plasma levels of different pro- and anti-inflammatory mediators in patients with sickle cell disease during crisis and at steady state, as compared to age-matched, healthy participants (A) MCP-1, (B) MIP-1α, (C) Eotaxin, and (D) IL-10. MCP-1 indicates monocyte chemoattractant protein 1; MIP-1α, macrophage inflammatory protein 1α; IL, interleukin.
Plasma levels of different growth factors including vascular endothelial growth factor (VEGF), fibroblast growth factor (FGF), or hepatocyte growth factor (HGF) in patients with SCD showed a distinct rise (P < .01), either during painful crisis or at steady state, with about 3-fold increase above that of healthy, age-matched control group for VEGF, FGF, or HGF (Figure 3A–C). Plasma levels of interferon γ (IFN-γ) showed a trend toward slight elevation during painful crisis that decreased during steady state but was not statistically significant compared to the levels in healthy, age-matched control group (Figure 3D). These data suggested a compensatory and sustained increase in growth factors in patients with SCD, perhaps to promote neovascularization as a response to vascular occlusion and hypoxia-mediated upregulation of growth factor production.
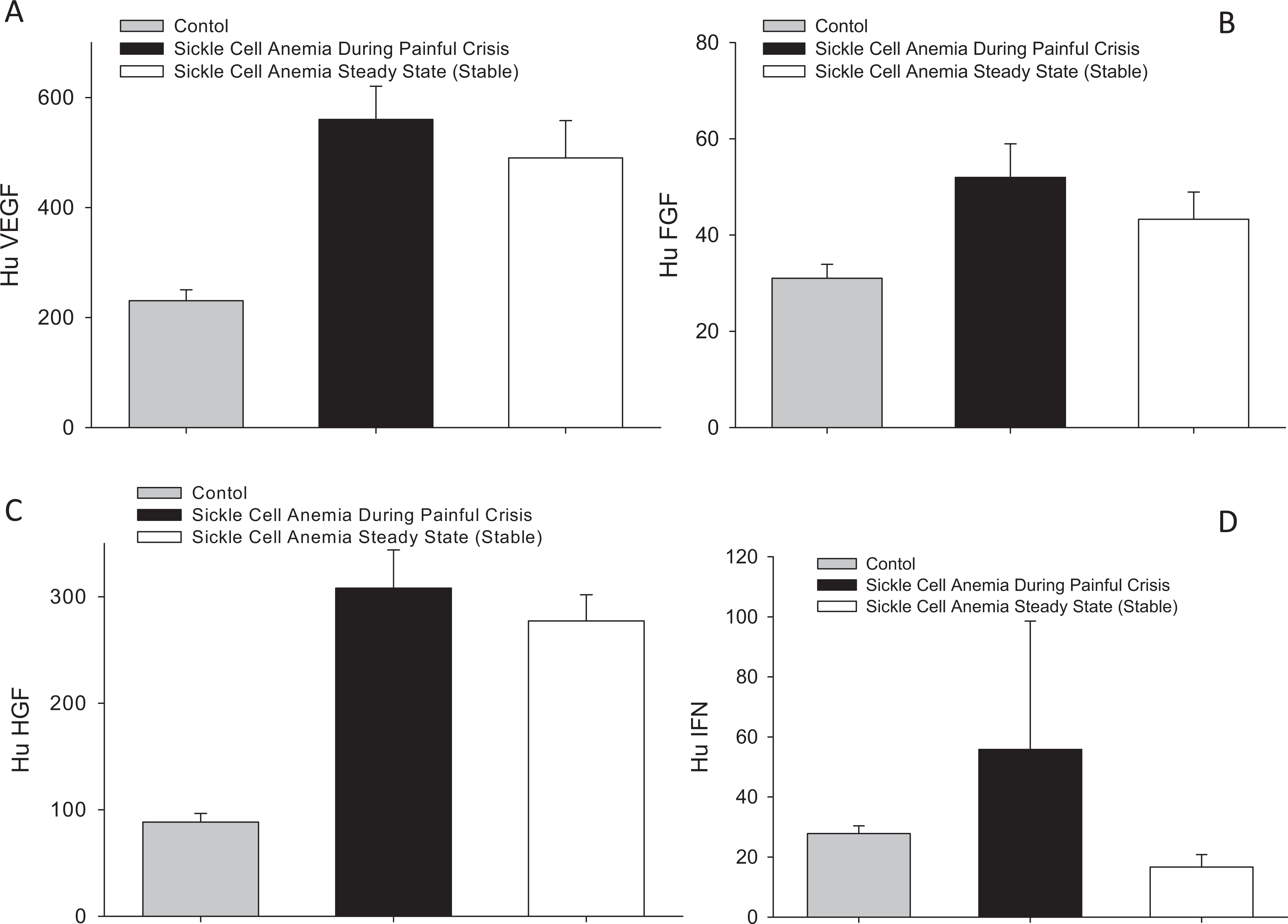
Plasma levels of different growth factors in patients with sickle cell disease during crisis and at steady state, as compared to age-matched, healthy participants (A) VEGF, (B) FGF, (C) HGF, and (D) IFN-γ. VEGF indicates vascular endothelial growth factor; FGF, fibroblast growth factor; HGF, hepatocyte growth factor; IFN-γ, interferon γ.
Plasma levels of
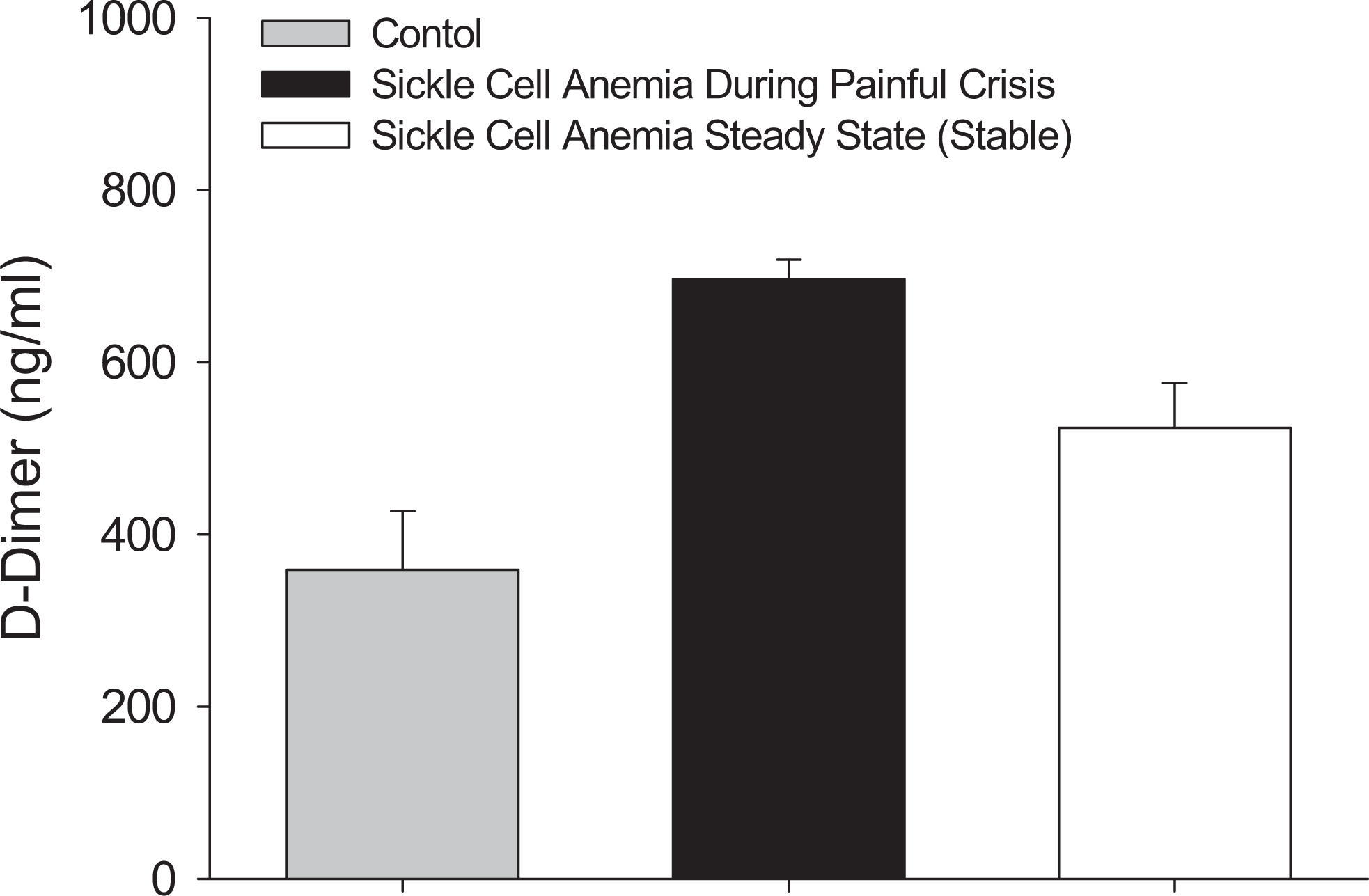
Plasma
Discussion
Inflammation, cell adhesion to vascular endothelium, and endothelial injury contribute to SCA vaso-occlusion. Although alterations in inflammatory cytokines and biomarkers have been related, reports have been conflicting, and a conclusive role for these molecules in the disease remains to be established. Data from this investigation revealed distinct upregulation of cytokine and chemokine production by at least 20- to 100-fold above age-matched control group, either during painful crisis or at steady state, suggesting sustained levels of proinflammatory stimuli despite a modest rise (2-fold) in natural, anti-inflammatory IL-10. This further suggested a high risk of chronic inflammation and its associated complications, such as promotion of prothrombotic state as evidenced from the increased
Based on the current biomarkers profile, clinical trials of anticoagulants and anti-inflammatory agents are warranted in patients with SCD. In that regard, low-molecular-weight heparin (LMWH) demonstrated effective release of endothelial tissue factor pathway inhibitor (TFPI) and increased NO production, which would overcome increased procoagulant activities and vasodilate vascular beds. 18 A recent study showed correlation of oxidative stress and inflammatory markers with the severity of sickle cell nephropathy. 19 Double-blind, randomized clinical trial with the LMWH tinzaparin in patients with SCD demonstrated rapid resolution of painful crisis. 1 Furthermore, studies with LMWH in other human participants suggested anti-inflammatory efficacy, 20 which might benefit patients with SCA.
Our investigation provided evidence of changes in the levels of inflammatory, proangiogenesis, and prothrombotic indicators in patients with SCD during painful crisis and at steady state as compared to age-matched, healthy human participants.
Footnotes
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The authors received no financial support for the research, authorship, and/or publication of this article.