
Abstract
The aim of this study was to review the literature for the genetic mutations causing inherited factoe XIII (FXIII) deficiency in patients from Iran, where the consanguineous marriage is common. Data were collected from 30 patients (18 males and 12 females) with FXIII deficiency, from 26 unrelated families. Data of mutation analysis were obtained from 2 previously published studies. A total of 7 mutations consisting of 5 new mutations and 2 previously reported mutations were identified. Of the 5 novel missense mutations, 2, Arg77His and Trp187Arg, were the most common in Iranian FXIII-deficient patients. In regions like Iran with high rate of consanguineous marriages, the identification of common mutations in disease like severe FXIII deficiency increases the capacity to make a precise screening and diagnosis assays to screen and diagnose families with high risk of FXIII deficiency for prevention of clinical complications in them.
Keywords
Introduction
Factor XIII (FXIII) is a zymogene which, converted into its active form (FXIIIa), promotes clot stability catalyzing covalent (ϵ-[γ-glutamyl] lysine) bonds between fibrin monomers and also by cross-linking fibrin with other proteins in the final stages of the blood coagulation pathways.1,2 Factor XIII is a transglutaminase consisting of 2 catalytic A subunits and 2 noncatalytic B subunits that stabilize FXIII-A.1,2
The gene coding for A subunit is located on chromosome 6p24-25. It spans 160 kb and consists of 15 exons separated by 14 introns. The A subunit constitutes the catalytic moiety composed of 731 amino acids and consists of 5 domains: an activation peptide (residues 1-37), a β-sandwich (residues 38-183), a catalytic core region (residues 184-515), and 2 β-barrels (barrel 1, residues 516-627 and barrel 2, residues 628-731). 3 The gene coding for B subunit is located on chromosome 1q31-32. It spans 28 kb and consists of 12 exons and 11 introns. The B subunit is composed of 640 amino acids forming 10 tandem repeats (sushi domains). 4
Congenital FXIII deficiency is a rare autosomal recessive coagulation disorder affecting approximately 1 out of 1 to 3 million people that is characterized by a tendency for spontaneous bleeding and impaired wound healing, intracranial hemorrhages, habitual miscarriage, and bleeding from umbilical cord and circumcision sites. 1
Deficiency of both the FXIII-A and B subunits has been described. However, in the majority of cases, inherited autosomal recessive FXIII deficiency is due to mutations in the gene coding for the A subunit, which result in severe bleeding. 5 Based on data obtained from FXIII registries and database (www.f13-database.de; www.hgmd.cf.ac.uk, and www.med.unc.edu/isth/mutations), more than 80 different mutations in FXIII-A gene have been reported. The FXIII-A gene is also known to be highly polymorphic, 5 polymorphisms were reported to have functional effects. 6 At variance, only 4 cases of FXIII-B deficiency due to mutation on the FXIII-B gene were reported so far resulting in mild-to-moderate bleeding.7,8
It is now well documented that FXIII deficiency is more prevalent in regions with high consanguinity where the probability of inheriting 2 abnormal alleles rises, raising in turn the possibility of expressing the disease otherwise rare.4,9 The aim of this study was to review the literature for genetic mutations causing the inherited FXIII deficiency in Iran, where consanguineous marriages are customary used.
Materials and Methods
Data of mutation analysis in this study were obtained from 2 previously published studies from Peyvandi et al 10 and Trinh et al. 11 A total of 30 Iranian patients (18 males and 12 females) from 26 unrelated families with FXIII deficiency were studied.
Results
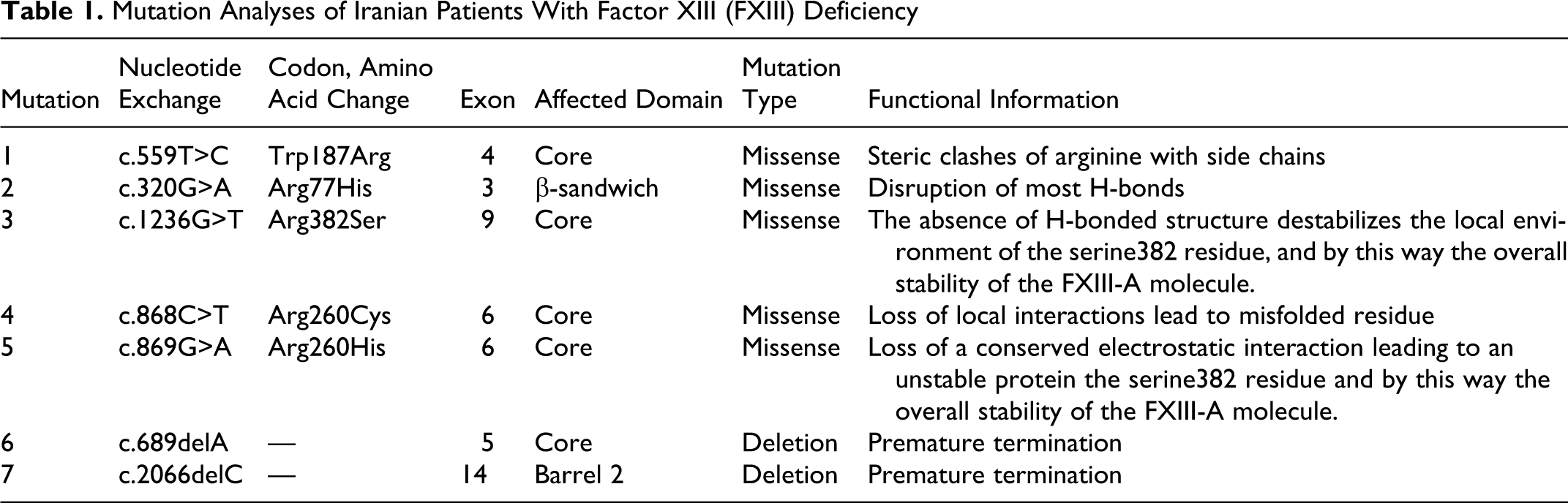
Seven causative mutations summarized in Table 1 were reported in 30 Iranian FXIII-deficient patients.
Mutation Analyses of Iranian Patients With Factor XIII (FXIII) Deficiency
c.320 G>A
This mutation reported in 5 patients is located in exon 3 of β-sandwich domain. It corresponds to Arg77His substitution at residue 77. The arginine 77 side chain takes part in an extended hydrogen bond (H-bond) network, which in the case of the histidine substitution to arginine causes no equivalent H-bonded network and low stability of the FXIII-A subunit. 10
c.559 T>C
This mutation reported in 20 patients is located in exon 4 coding for the core domain, and it corresponds to Trp187Arg change at residue 187. Substitution of arginine to tryptophan in this position causes steric clashes between the arginine atom and the surrounding atoms from the neighboring residues that form the hydrophilic cavity and result in some localized rearrangement in neighboring residues to minimize the result and steric clashes. These rearrangements may cause destabilization of this region within the FXIII-A subunit. 11
c.689 delA
This single nucleotide deletion in exon 5 coding for the core domain causes a premature stop. It was reported in 1 patient. 10
c.868 C>T
This mutation reported in 1 patient is located in exon 6 coding for the core domain. It corresponds to Arg260Cys. The substitution of cysteine to arginine at residue 260 leads to complete absence of H-bond network that results in abnormal folding, instability of the protein, and can hinder the dimerization of FXIII-A subunit. 10
c.869 G>A
This mutation, reported in 1 patient, is located in exon 6 coding for the core domain. It corresponds to Arg260His substitution at residue 260. The effect of this mutation is that only 1 strong H-bond involved in keeping the 2 subunits together is left. 10
c.1236 G>T
This mutation, reported in 1 patient, is located in exon 9 coding for the core domain. It corresponds to Arg382Ser substitution. This substitution at residue 382 in contrast to normal condition leaves only 1 weak H-bond which could be the cause for the destabilization of FXIII-A subunit. 10
c.2066 delC
This single nucleotide deletion in exon 14 coding for the barrel 2 domain causes a premature stop. This mutation was reported in 1 patient. 10 A schematic presentations of the mutations analyzed is shown in Figure 1.
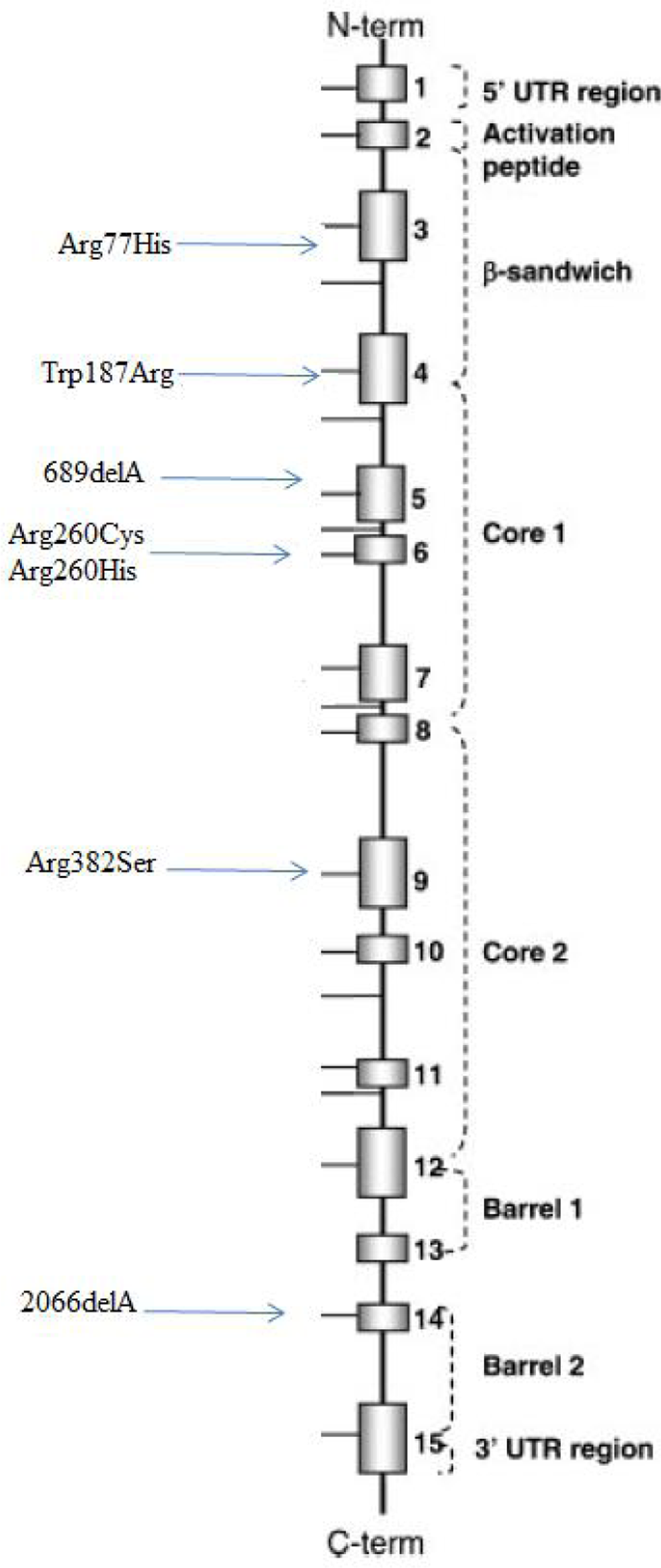
Schematic presentations of the factor XIII (FXIII) mutations in Iran patients.
Discussion
Causative mutations have been found both in FXIII-A and FXIII-B genes. Most of mutations reported within the FXIII-A gene are missense/nonsense mutations. Missense mutations resulted in defective folding and instability of FXIII-A subunit. Nonsense mutations and small deletions led to frameshift and consequently to premature termination protein synthesis.1
Of the 5novel missense mutations, 2, Arg77His and Trp187Arg, were the most common mutations identified in Iranian FXIII-deficient patients. Two missense mutations Arg260Cys and Arg260His were reported previously in Japanese and Indian patients, respectively.12,13 Four missense mutations resulted in the substitution of arginine by histidine, cysteine, or serine, which destroys or impairs H-bond network and destabilization energy and finally instability of the FXIII-A subunit. Steric clashes of arginine with side chains prevalent in missense mutation Trp187Arg cause the rearrangement of the local residues and instability of the FXIII-A subunit. Premature termination of protein synthesis in small deletion mutations can cause severe decrease FXIII levels.
Iran is a country with about 70 million people that has a traditional culture as well as consanguineous marriages that can cause high rate of congenital diseases.
In conclusion, in regions like Iran with high rate of consanguinity marriages, the identification of common mutations in the prevalence of FXIII deficiency increases the capacity to make a precise screening and diagnosis assays to screen and diagnose families and patients with FXIII deficiency for prevention of clinical complications in them.
Footnotes
Acknowledgments
We thank Shirin Parand at the Hematology Research Center, Nemazee Hospital, and Shiraz for editorial assistance.
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The authors received no financial support for the research, authorship, and/or publication of this article.