
Abstract
Aims:
Hydrogen sulfide (H2S) protects against ischemic and inflammatory injury following myocardial ischemia via induction of microRNA (miR)-21. We sought to determine whether H2S attenuates ischemic heart failure with reduced ejection fraction (HFrEF) and interrogate the role of cofilin-2, a target of miR-21, in this protective process.
Methods and Results:
Adult male mice underwent myocardial infarction (MI) by coronary artery ligation after baseline echocardiography. Following MI, mice were treated with Na2S (100 μg/kg/day; intraperitoneal [IP]) or saline up to 28 days. End-diastolic pressure, measured by Millar catheter, was significantly increased (P < .05 vs sham) at 3 days post-MI in the saline group, which was attenuated with Na2S. Left ventricular (LV) fractional shortening decreased significantly at 28 days post-MI in the saline group but was preserved with Na2S and LV infarct scar size was smaller in Na2S group as compared to control. Apoptotic signaling, measured by Bcl-2/Bax ratio, was significantly increased in the saline group but was mitigated with Na2S. Survival rate was 2-fold higher in Na2S group compared to saline control (P < .05). Proteomic analysis and Matrix-Assisted Laser Desorption/Ionization-Time of Flight (TOF)/TOF tandem mass spectrometry identified significant changes in proapoptotic cofilin-2 expression, a specific target of miR-21, between saline- and sodium sulfide -treated mice at 28 days post-MI. Western blot analysis confirmed a significant increase in cofilin-2 after MI, which was suppressed with Na2S treatment. Chronic Na2S treatment also attenuated inflammasome formation and activation leading to reduction of maladaptive signaling.
Conclusion:
Na2S treatment after MI preserves LV function and improves survival through attenuation of inflammasome-mediated adverse remodeling. We propose H2S donors as promising therapeutic tools for ischemic HFrEF.
Introduction
Dysregulation of endogenous concentrations of hydrogen sulfide (H2S) has been demonstrated in several health-related conditions ranging from heart failure to nervous system disorders and inflammatory syndromes. 1 Hydrogen sulfide is a gasotransmitter that tightly regulates physiological processes including promoting angiogenesis in the context of cardiac ischemia, attenuating myocardial infarction (MI) following ischemia and reperfusion injury, and exhibiting antiapoptotic effects in cardiomyocytes. 1 Studies have demonstrated a protective role of H2S against oxidative stress in ischemic injury and anti-inflammatory effects in the setting of myocardial ischemia and reperfusion injury. 2 Importantly, a recent phase I clinical trial with SG1002, a novel orally active H2S releasing prodrug, demonstrated its safety and tolerability in heart failure patients and control patients. 3 The study also indicated that SG1002 was able to restore H2S levels in HF patients while also improving their nitric oxide levels and decreasing brain natriuretic peptide (BNP), a biomarker of HF.
MicroRNAs (miR) are endogenous ∼22 nucleotides single-stranded RNAs that bind to the 3’ region of mRNA and regulate gene expression at the posttranscriptional level. 4 Our laboratory previously demonstrated miR-21 played a cardioprotective role after acute myocardial infarction (AMI) by decreasing myocardial infarct size and attenuating apoptosis. 5 Furthermore, we have shown in a murine model of AMI that exogenous H2S protected the myocardium by inhibiting the formation and activation of the NLRP3 inflammasome through a novel signaling pathway dependent upon miR-21. 6 However, little data exist regarding the cardioprotective effects of H2S in ischemic heart failure with reduced ejection fraction (HFrEF), which remains a major post-MI complication. 7,8 Additionally, the downstream effectors of the H2S-miR-21 axis are poorly defined.
In this study, we examined exogenous H2S treatment in ischemic HFrEF using a murine model of permanent left anterior descending coronary artery ligation. We interrogated the protective effects of H2S against inflammasome formation/activation, apoptosis, and fibrosis. To understand the downstream signaling of H2S in ischemic cardiomyopathy, we took advantage of proteomic analysis and determined that cofilin-2 may be a target of the H2S signaling pathway. Cofilin-2 is a 19-kDa protein belonging to a family of actin-binding proteins that participates in the disassembly of the sarcomeric protein actin. 9 Recently, hyperphosphorylated cofilin-2 has been shown to form pre-amyloid oligomer aggregates under pathophysiologic states in the human myocardium, which disrupt its structure and function resulting in a form of idiopathic dilated cardiomyopathy. 9 Furthermore, cofilin-2 expression can be detrimental through its proapoptotic effect by inducing cytochrome c release from mitochondria. 10
Materials and Methods
Animals
Adult male C57BL mice were purchased from The Jackson Laboratories. The experiments involving animals were performed in accordance with the guidelines on humane use and care of laboratory animals for biomedical research published by the National Institutes of Health (No. 85-23, revised 1996). The animal protocol was approved by the Institutional Animal Care and Use Committee at Virginia Commonwealth University.
Drugs and Chemicals
Sodium sulfide (Na2S) was purchased from Sigma-Aldrich. Primary antibodies against Cofilin-2, Bcl-2, Bax, and galectin-3 were purchased from Santa Cruz Biotechnologies. Anti-GAPDH was purchased from Cell Signaling Technology. Antiapoptosis Speck (ASC)-like protein and anticardiac actin were purchased from Sigma-Aldrich. Secondary antibody Anti-rabbit Alexa Fluor 594 and 488–conjugated, 4′,6-diamidino-2-phenylindole (DAPI), and Slow Antifade were purchased from Invitrogen. Caspase-1 fluorogenic substrate Ac-Tyr-Val-Ala-Asp-7-amino-4-methyl coumarin (Ac-YVAD-AMC) was purchased from Enzo Life Sciences. Radioimmunoprecipitation assay (RIPA) buffer was purchased from Cell Signaling Technology. Masson trichrome staining kit was purchased from Sigma-Aldrich.
Myocardial Infarction Protocol
We used a model of permanent left coronary artery ligation in order to investigate ventricular remodeling and cardiac function following MI secondary to severe ischemia. Permanent coronary occlusion induces robust myocardial damage that translates into long-term ventricular adverse remodeling, recapitulating most of the pathophysiological features of patients with ischemic heart failure.
The methodology of MI was described previously. 11 In summary, the animals were orally intubated under anesthesia (pentobarbital 50-70 mg/kg) and placed on positive-pressure ventilation. A left thoracotomy was performed at the fourth intercostal space, and the left anterior descending coronary artery was identified and permanently ligated with a 7.0-silk ligature. Sham-operated mice were subjected to left thoracotomy without permanent arterial ligation. The surgeon was blinded to treatment allocation in the MI groups.
Experimental Groups (In Vivo)
Immediate Na2S Treatment
C57BL wild-type mice were randomly assigned to either saline control (0.2 mL daily, IP) or Na2S (100 μg/kg daily, IP) immediately following left anterior descending (LAD) ligation up to 28 days (Figure S1). This therapeutic dose of Na2S has been well documented by our group and others in the literature.
Late Na2S Treatment
To mimic a clinical model of ischemic HFrEF, we randomly assigned a group of adult mice with left ventricular (LV) FS <25% at 3 days post-MI to either saline control (0.2 mL daily, IP) or Na2S treatment (100 μg/kg daily, IP) up to 28 days post-MI (Figure S1).
Survival
Survival rate was established by taking into account the animals that survived the MI surgical protocol and were able to breathe independently during recovery until the date of death.
Echocardiography
Echocardiography was performed using the Vevo2100 imaging system (VisualSonics Inc, Toronto, Ontario, Canada) before surgery (baseline) and on days 3, 7, and 28 post-MI. Isoflurane 1.5% to 3% in oxygen vaporizer system was used for anesthesia and the procedure was performed as previously described. 12 To measure LV end-diastolic diameter and end-systolic diameter (LVEDD and LVESD, respectively) and anterior wall diastolic thickness (ADWT). Left ventricular fractional shortening was calculated using the following formula: (LVEDD-LVESD)/LVEDD × 100. The sonographers were blinded to treatment allocation at all time points.
Left Ventricular Contractile Function and Hemodynamics
After anesthetizing the animal (pentobarbital 50 mg/kg IP), a micro-tip pressure-volume catheter transducer (model SPR-1045; Millar Instruments, Inc) was advanced into the LV cavity through the right carotid artery. A short period of stabilization was allowed before continuously recording signals using a MPVS-300 system (Millar Instruments) coupled with a Powerlab 8/30 converter (ADInstruments, Inc). The data were stored and displayed on a computer. Left ventricular systolic and end-diastolic pressures, maximal slope of systolic pressure increment (+dP/dtmax), diastolic pressure decrement (−dP/dtmax), heart rate, and aortic blood pressure were recorded continuously throughout the procedure. The surgeon was blinded to treatment allocation.
Proteomic Analysis by 2D-Gel, Spot Picking and Digestion, and MALDI-TOF/TOF
The heart samples were collected and flash frozen in liquid nitrogen for proteomic analysis. The 2-dimensional differential in-gel electrophoresis (2D-DIGE) and mass spectrometry protein identification were performed by Applied Biomics, Inc as previously reported. 13
Typhoon Trio scanner (Amersham) was used to scan the gels after sodium dodecyl sulfate–polyacrylamide gel (SDS-PAGE) electrophoresis, and the images were analyzed using Image QuantTL software (GE-Healthcare), before being subjected to in-gel as well as cross-gel analysis using DeCyder software version 6.5 (GE Healthcare). The ultimate alteration in expression of various proteins was obtained from in-gel DeCyder software analysis. Quantitative comparisons were then made between 2 individual samples for each of the 3 possible combinations. A pair-wise volume was calculated for each protein spot and used to determine relative protein expression. To render relevant data presentation simpler, we only reported the ratios of comparisons between MI and MI + Na2S.
Spots were identified by Ettan Spot Picker (GE-Healthcare) according to DeCyder software analysis. Afterward, the protein spots of choice underwent digestion by in-gel trypsin, extraction of peptides, and desalting followed by Matrix-Assisted Laser Desorption/Ionization-Time of Flight (MALDI-TOF)/TOF in order to identify the proteins. Briefly, mass spectra (MS) of the peptides from each sample were obtained using Applied Biosystems Proteomics Analyzer. Between 10 and 20 of the most abundant peptides in each sample were fragmented further and subjected to tandem mass spectrometry (MS/MS) analysis. Fingerprint mass mapping (based on MS spectra) and peptide fragmentation mapping (based on MS/MS spectra) were used to identify proteins. Combined MS and MS/MS spectra were submitted for database search using GPS Explorer software equipped with the MASCOT search engine to identify proteins from primary sequence databases.
Western Blot Analysis
Isolated LV samples (n = 3/group) were homogenized, sonicated, and centrifuged at 10 000g for 15 minutes at 4 °C. Total proteins (100 µg) from each sample were separated by SDS-PAGE on 10% acrylamide gels, transferred onto a nitrocellulose membrane, and blocked with 5% nonfat dry milk in Tris-buffered saline. Membranes were incubated overnight with antibodies to cofilin-2 (1:1000), apoptotic proteins Bcl-2 and Bax (1:1000), galectin-3 (1:1000), GAPDH (1:5000). Membranes were developed with enhanced chemiluminescence and subsequently exposed to X-ray film. Protein levels were quantified using densitometry (Figure S2).
Real-Time PCR
Following 28 days after MI, transcript levels of transforming growth factor β, intercellular adhesion molecule 1 (ICAM-1), tumor necrosis factor (TNF), interleukin-1 β, and Apoptosis regulator BAX were quantified by real-time PCR performed using PrimePCR Arrays in the CFX96 Touch Real-Time PCR Detection System (Bio-Rad Laboratories) using the SSOAdvance Universal SYBR Green Super mix for Real-Time PCR (Bio-Rad Laboratories). All the samples were processed in triplicates according to the manufacturers’ recommended conditions. The cycling conditions were as follows: 95 °C for 30 seconds, 95 °C for 15 seconds, 60 °C for 30 seconds, 40 cycles of 95 °C for 15 seconds, and 60 °C for 30 seconds.
Histology
Transverse sections of the median third of the LV were fixed in 10% formalin for at least 48 hours, embedded in paraffin, and sectioned into 5-µm sections. Heart sections were stained with Masson trichrome to examine scar formation within the LV (Richard-Allan Scientific Masson Trichrome Kit; Thermo Scientific). Fibrotic area was computed using computer morphometry ImageJ (National Institutes of Health) and expressed as percentage of LV.
Following manufacturer’s instructions, terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay was used to assess myocardial apoptosis in paraffin-embedded heart sections (Click-it Plus TUNEL Assay; Invitrogen). Cell counterstaining was performed using DAPI (1:1000) for 5 minutes, and the slides were cover-slipped with SlowFade Antifade. Microphotographs of the infarcted and border zones were obtained using an IX70 microscope and the MagnaFire 1.1 software (both Olympus) using a ×20 objective (×200 magnification). Color composite images were generated and quantified using ImageJ software. Apoptosis was expressed as percentage of TUNEL-positive cells over total nuclei counted.
Measurement of the Inflammasome in the Heart
At the conclusion of the MI protocol, heart sections (3-µm thick slices) were prepared after fixing the tissue in formalin and embedding in paraffin. Following these initial steps, the heart section underwent deparaffinization and rehydration with serial washes in xylene and decreasing concentrations of ethanol. Antigen retrieval was attained with 0.01 mol/L citrate buffer (pH 6.0) for 20 minutes, and then the slides were blocked for 1 hour with 5% normal donkey serum in phosphate-buffered saline (PBS). A double immunofluorescence technique was used for characterization of cardiomyocyte-specific expression of the aggregates of the scaffold protein ASC, which is indicative of inflammasome formation. Primary antibody for ASC (1:50) was used to incubate the slides overnight at 4 °C. The slides were then exposed to Anti-rabbit Alexa Fluor 594–conjugated secondary antibody (1:100) for 4 hours at room temperature, after which they were incubated overnight with cardiac actin primary antibody (1:200) at 4 °C, before the application of Alexa Fluor 488–conjugated secondary antibody (1:100) for 4 hours at room temperature. Cell counterstaining was performed using DAPI (1:1000) for 5 minutes, and the slides were cover-slipped with SlowFade Antifade. Slides with nonspecific immunoglobulin G were run in parallel as negative control. Images were acquired with an IX70 microscope and the MagnaFire 1.1 software (both Olympus) using a ×40 objective (×400 magnification). Color composite images were generated with ImageJ software.
ASC assessment in the infarct region (area at risk) was performed in a blinded fashion by 2 independent investigators, and the expression was measured using a semi-quantitative scale: 0 reflecting no expression; 1+ reflecting minimal expression demonstrating either few aggregates (<1 per high-power field) or mild diffuse stain with no aggregates; 2+ reflecting moderate expression demonstrating either 1 to 5 aggregates per high-power field or diffuse stain with few aggregates; 3+ reflecting diffuse intense staining with many cytoplasmic aggregates (>5 per high-power field).
Caspase-1 Activity in the Heart
Another subgroup of mice was euthanized 72 hours post-MI (n = 4-6 per treatment group). After procurement, the hearts were rinsed in PBS before being flash-frozen in liquid nitrogen and stored in a −80 °C freezer. Tissue caspase-1 activity was assessed by utilizing a fluorogenic substrate Ac-YVAD-AMC specific for caspase-1. The tissue was homogenized using RIPA buffer that contains a cocktail of protease inhibitors and then centrifuged at 16 000 rpm for 20 minutes, and 50 µg of protein per sample were used for the assay. One hour later, measurement of fluorescence was performed and the results were expressed as arbitrary fluorescence units produced by 1 µg of sample per minute (fluorescence/µg per minute). Final calculation was based upon fold change compared with the sham hearts.
Statistical Analysis
Data results are calculated as group means ± standard error. Differences in echocardiography, hemodynamics, and myocardial fibrosis were analyzed using 2-way ANOVA. Western blot results were analyzed using 1-way ANOVA or t test with post hoc Tukey test when appropriate. Discrete variables between groups were evaluated using the χ2 test. Kaplan-Meier analysis was used to estimate differences in survival from the point of recovery following surgery to the time of death. Statistical significance was set at P < .05.
Results
Na2S Improves Survival in MI-Induced Heart Failure
A total of 75 mice were enrolled in this study. The immediate sodium sulfide (Na2S)-treated group had 10 of 21 mice survive at 28 days post-MI demonstrating a higher survival rate in comparison to 8 of 33 that survived in the saline-treated group (P < .05) as shown in Figure 1A.
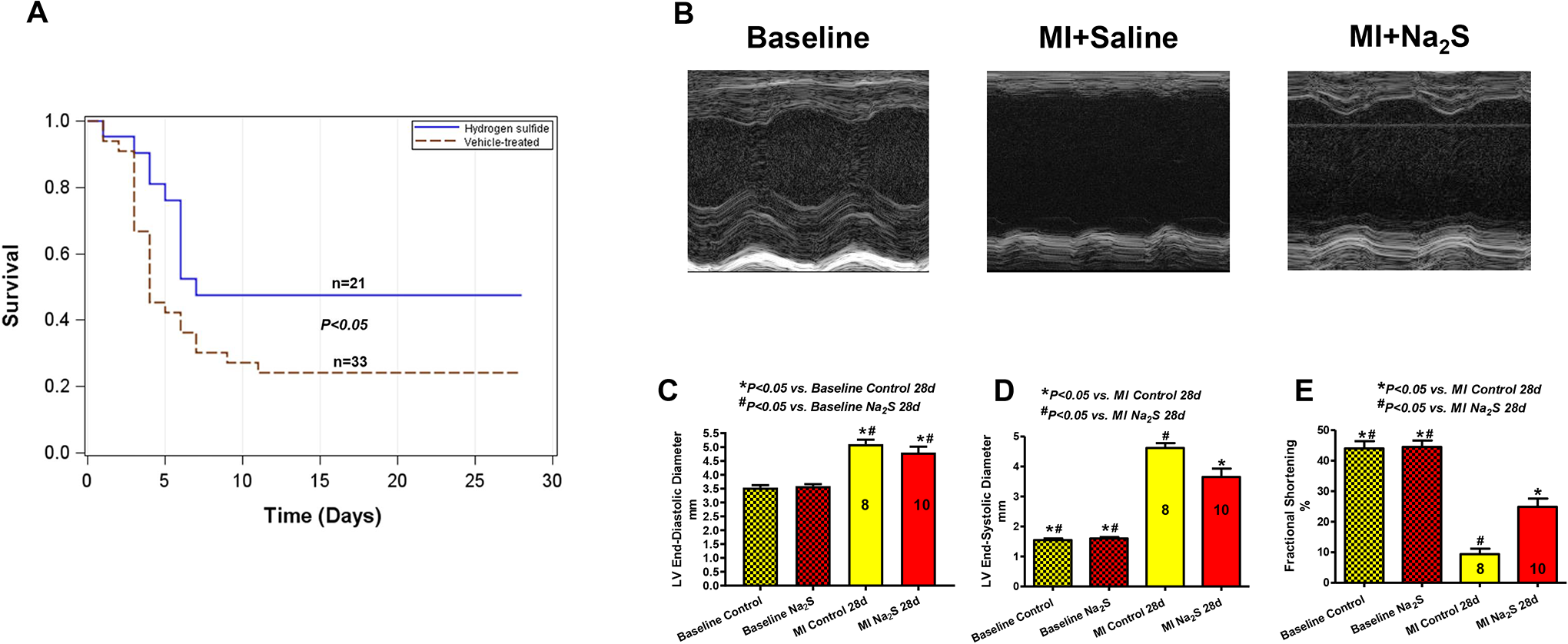
A, Administration of Na2S immediately following LAD occlusion caused a significant 2-fold increase in survival rate at 28 days compared to saline-treated mice. B, M-mode representative images of different groups, at baseline and 28 days post-MI. C-E, LVEDD, LVESD, and FS at baseline and at 28 days post-MI. Na2S treatment significantly decreased LVESD as compared to saline (# P < .05), which translated into significant preservation in LV fractional shortening. LAD indicates left anterior descending; LV, left ventricle; LVEDD, LV end-diastolic diameter; LVESD, LV end-systolic diameter; MI, myocardial infarction.
Chronic Na2S Therapy Starting at the Time of Ischemia Attenuates Left Ventricular Adverse Remodeling Up to 28 days Following MI
Representative M-mode images at baseline and 28 days post-MI in mice immediately treated with saline or Na2S are shown in Figure 1B. Baseline fractional shortening was 44% ± 2%. Even though both saline- and Na2S-treated mice had dilatation in LVEDD as compared to baseline (P < .05), the LVESD in Na2S group was less compared to saline treatment (Figure 1C and D; Table 1). At 28 days, LV systolic function (ie, LVFS) was significantly preserved in Na2S group as compared to saline control (FS: 25% ± 3% vs 9% ± 2%, respectively, P < .05, Figure 1E; Table 1). This observation also reflected less anterior wall thinning with Na2S therapy (AWDT: 0.9 ± 0.15 mm) as compared to saline control (AWDT: 0.42 ± 0.19 mm, P < .001; Table 1). Interestingly, hemodynamic assessment at 3 days post-MI showed that the Na2S group had lower EDPVR slopes compared to saline group (P < .05, Figure 2A and B; Table 2), suggesting less diastolic dysfunction from MI-induced LV remodeling.
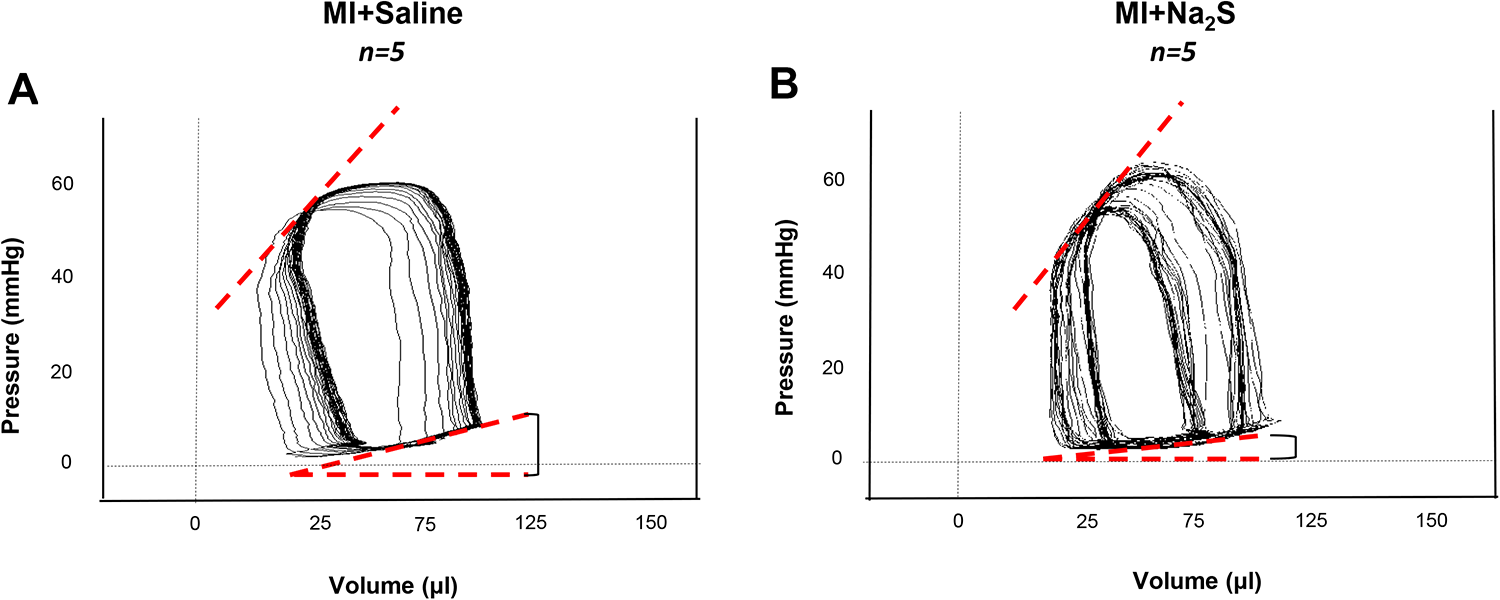
Left ventricular Millar catheterization. Representative images of pressure/volume loop recordings of (A) vehicle and (B) Na2S-treated hearts 3 days following AMI. Sodium sulfide -treated mice showed lower EDPVR slopes compared to vehicle treated mice (1.4 ± 0.8 vs 6.3 ± 1.1, respectively; P = .01; N = 5/group), reflecting better LV compliance with H2S treatment. AMI, acute myocardial infarction; LV, left ventricle; Na2S, sodium sulfide.
Echocardiographic Parameters.

Abbreviations: FS, fractional shortening; LVEDD, left ventricle end-diastolic diameter; LVESD, left ventricle end-systolic diameter; AWTD, anterior wall thickness in diastole.
a P < .05 vs MI Na2S 28 days.
b P < .05 vs MI control 28 days.
Intraventricular Catheterization Parameters at 3 Days Following MI.
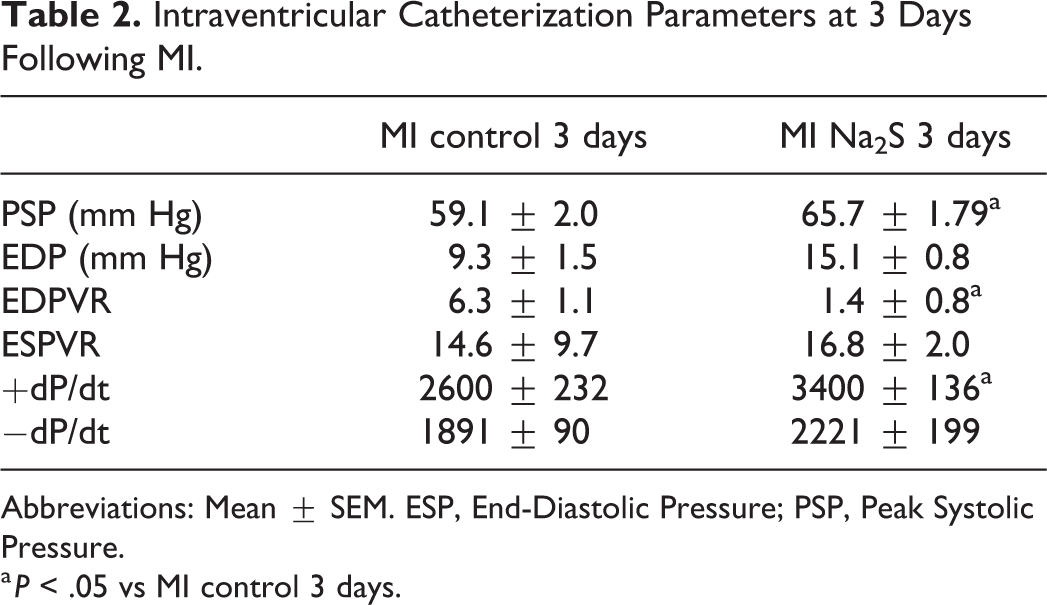
Abbreviations: Mean ± SEM. ESP, End-Diastolic Pressure; PSP, Peak Systolic Pressure.
a P < .05 vs MI control 3 days.
Histologically, myocardial fibrosis (%LV) was higher in saline (33% ± 2%) compared with Na2S-treated mice (22% ± 3%, P < .05, Figure 3A and B) 28 days post-MI. Pro-inflammatory and fibrotic cytokines TGF-β, ICAM-1, TNF, and IL-1 were increased in the saline-treated myocardium as compared to sham, but their levels were blunted in Na2S-treated group (P < .05, Figure 3C). Moreover, MI was associated with a 60-fold increase in pro-fibrotic Galectin-3, which was significantly attenuated with Na2S (Figure 3D).

A-B, Representative heart sections showing a significant reduction in LV scar with Na2S treatment at 28 days post-MI. C-D, When compared to saline treatment, Na2S significantly attenuated at 28 days the surge of myocardial TGF-β, ICAM-1, TNF, IL-1β, and BAX as well as Galectin-3 associated with MI. ICAM-1 indicates intercellular adhesion molecule 1; IL-1β, interleukin-1-1β; LV, left ventricle; MI, myocardial infarction; TGF-β, transforming growth factor; TNF, tumor necrosis factor.
Na2S Therapy Reduces Apoptosis Following MI
Apoptosis in the infarcted area and the border zone was quantified by TUNEL Assay. The apoptotic rate was significantly increased following MI at 28 days. Treatment with Na2S significantly reduced apoptotic nuclei following MI at 28 days (P < .05, Figure 4).

A-B, Representative heart sections showing a significant reduction in apoptotic nuclei following Na2S treatment at 28 days post-MI. (C-D) when compared to saline treatment, Na2S significantly attenuated at 28 days the surge of myocardial TGF-β, ICAM-1, TNF, IL-1β, and BAX as well as galectin-3 associated with MI. ICAM-1 indicates intercellular adhesion molecule 1; IL-1β, interleukin-1-1β; MI, myocardial infarction; TGF-β, transforming growth factor; TNF, tumor necrosis factor.
Cardiac Bcl-2, a pro-survival protein, decreased following MI as compared to baseline (P < .05, Figure 5A and B). Pro-apoptotic cardiac Bax expression was significantly increased with MI (P < .05 vs baseline), and this increase was mitigated with Na2S therapy (P < .05 vs MI) at day 28 post-MI. As a result, the Bcl-2/Bax ratio decreased following MI as compared to baseline (P < .05), but this decline was significantly attenuated with Na2S treatment (Figure 5C and D). These results support the notion that Na2S reduced the apoptotic signaling cascade that accompanied MI-induced HFrEF.

Western blot analysis demonstrating a significant decrease in Bcl-2/Bax ratio at 28 days post-MI, which was significantly preserved Na2S treatment. MI, myocardial infarction.
Na2S Treatment Attenuates Formation and Activation of Cardiac NLRP3 Inflammasome
Treatment with Na2S starting at the onset of MI blunted cardiac NLRP3 inflammasome formation as evidenced by a decrease in ASC aggregates in the zone bordering the infarct (by 37%, P < .05; Figure 6A and B) and caspase-1 activity (Figure 6C) at 3 days following MI.

A-B, Sodium sulfide. treatment immediately following permanent LAD occlusion attenuated the increase in ASC in the heart 3 days later. C, Correlating with ASC staining and inflammasome activation, caspase-1 activity significantly increased with MI, and the increase was abrogated with Na2S therapy. ASC indicates Apoptosis Speck-like protein; LAD, left anterior descending; MI, myocardial infarction; Na2S, sodium sulfide.
Proteomic Analysis Identifies Cofilin-2 as a Downstream Target of Na2S Treatment in Ischemic Heart Failure
To understand the potential downstream effectors of Na2S in ischemic HF, we conducted proteomic analysis which revealed 46 protein spots with altered expression profiles between saline and Na2S-treated mice at 28 days post-MI (in excess of 1.5-fold change). Consequently, MALDI-TOF/TOF tandem mass spectrometry identified that there was an increase in cofilin-2 profile with MI that was blunted with Na2S therapy (Figure 7A). We used Western blot analysis to confirm a significant increase in cofilin-2 after permanent myocardial ischemia that was suppressed with Na2S treatment 28 days post-MI (Figure 7B and C). No significant differences in cofilin-2 expression were detected at both 3 and 7 days following MI (data not shown).

A, Scanned image of the 2-dimensional gels. Overlay image for gel comparison between 28 days post-MI heart from saline-treated C57BL mouse (green dye) and heart from C57BL mouse treated with Na2S (red dye). The circled and numbered protein spots (46 spots in total 90 spots with a cutoff ratio of 1.5 for alteration in protein expression among the treatment options) are subsequently isolated from the gels and identified by MALDI-TOF/TOF mass spectroscopy. Representative 3-dimensional DeCyder software integrated graph showing difference in Cofilin-2 (spot 79) between MI and MI + Na2S-treated mice. Proteomic analysis revealed a significant increase in proapoptotic cofilin-2 at 28 days post-MI, which was reversed with Na2S treatment. B-C, Western blot analysis confirmed alterations in cardiac cofilin-2 and proangiogenic VEGF expression profiles following MI, which were attenuated with Na2S. MALDI-TOF indicates Matrix-Assisted Laser Desorption/Ionization-Time Of Flight; MI, myocardial infarction; Na2S, sodium sulfide; VEGF, vascular endothelial growth factor.
Pro-angiogenic vascular endothelial growth factor (VEGF) expression was also preserved with Na2S therapy as compared to saline treatment (P < .05, Figure 7B and C).
Delayed Na2S Treatment (3 Days Post-MI) Reduces Left Ventricular Remodeling in Ischemic HFrEF
To mimic established ischemic HFrEF and dissect the acute protective effects of H2S at the time of AMI from its potential benefits if administered later during the course of post-MI HF, we initiated treatment with Na2S or saline in mice with LVFS <25% at 3 days post-MI. In this cohort, FS at treatment initiation was 11% ± 1%. Interestingly, at 28 days post-MI, FS in “delayed” Na2S treatment improved significantly to 20% ± 2% (P < .05, Figure 8A). In addition, at 7 days post-MI (4 days after treatment initiation), there was robust inflammasome formation as seen with ASC staining in the saline group, which was significantly attenuated with Na2S treatment (P < .05, Figure 8B and C). These data suggest that Na2S may participate in reverse remodeling to improve LV systolic function by blunting persistent sterile inflammation.
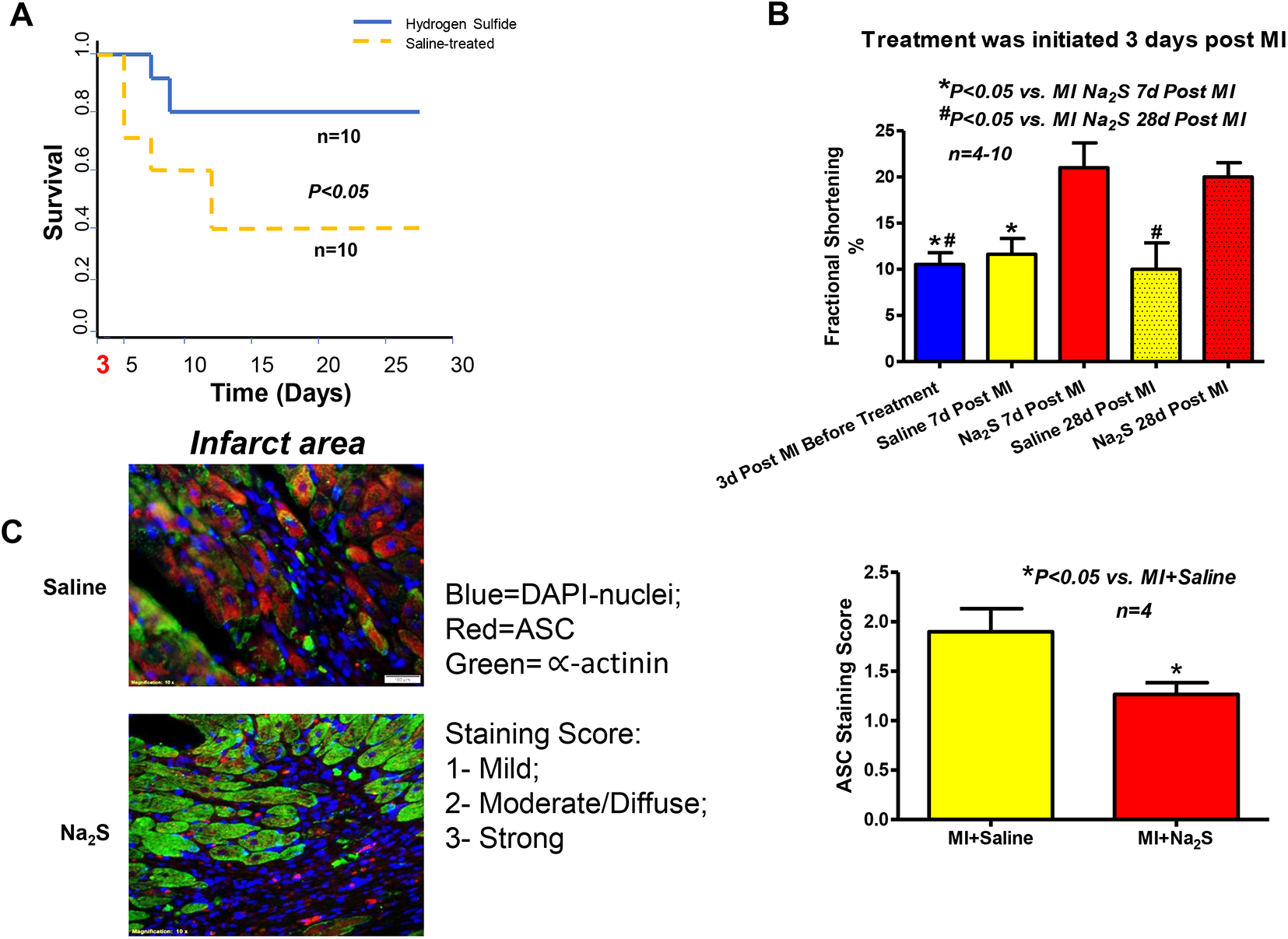
A, Sodium sulfide therapy in ischemic HFrEF (LVFS < 25%) starting at 3 days post-MI in surviving mice caused a significant 2-fold increase in survival rate at 28 days compared to saline-treated mice. B, Delayed Na2S treatment improved fractional shortening as compared to saline group. C, Antiapoptosis Speck staining score was significantly decreased with delayed Na2S treatment at 7 days post-MI compared to saline-treated mice. HFrEF indicates heart failure with reduced ejection fraction; MI, myocardial infarction; Na2S, sodium sulfide.
Discussion
Exogenous H2S treatment with Na2S donors can modulate several acute and chronic illnesses. In the cardiovascular system, H2S acts as a gasotransmitter to protect against ischemia, pressure overload, doxorubicin toxicity, inflammation, arterial contraction, blood vessel relaxation, and insulin release. 14 Moreover, endogenous H2S appears to be an effector of the cardioprotective nitric oxide-cGMP-PKG axis via phosphodiesterase-5 inhibitors 15 and the NO-independent guanylate cyclase activator, cinaciguat. 13 Previously, we have shown that exogenous H2S acutely protects against ischemia/reperfusion and inflammatory injury after MI via miR-21-dependent pathway. 6 However, the effects of chronic H2S therapy on sterile inflammation in ischemic cardiomyopathy and HFrEF remain unclear. Our present study has several novel findings: (1) Na2S therapy immediately after AMI or in established ischemic HFrEF attenuates adverse ventricular remodeling by reducing fibrosis, NLRP3 inflammasome activation, and apoptotic signaling, thus improving LV systolic function; (2) Proteomic analysis of downstream effectors of H2S therapy reveals that cofilin-2 may be a novel target. Although several mechanisms seem to be involved in mediating the effects of H2S therapy as mentioned above, whether these pathways are linked or function independently remains unclear and requires further investigation.
In response to ischemic myocardial injury, adverse left ventricular remodeling occurs resulting in inflammasome formation, oxidative stress, cellular death, and development of fibrosis. Especially critical is the role of inflammatory injury and its progression toward maladaptive signaling in the failing heart. 16 Previously, the progression of HF was believed to mainly be due to neurohormonal activation. 17 Recent studies, including the current findings, implicate inflammasome formation and activation as significant contributors to this pathophysiological process. 16 It has been shown that HF patients have diminished circulating levels of H2S in comparison to their age-matched control peers. 18 Given that ischemic HF patients exhibit H2S deficiency, supplementation with H2S donor therapy may represent a promising therapeutic approach, further supporting the recent findings with the novel H2S-releasing prodrug, SG1002 in a phase I clinical trial. 3
The cardioprotective mechanisms of endogenous and exogenous H2S have been shown to relate to the increased phosphorylation of Akt. 19 Furthermore, Akt, a pro-survival protein, was previously shown to positively regulate miR-21 that becomes deactivated during ischemia or hypoxia in cardiac myocytes. 20 From our previous study, we showed that Akt phosphorylation with Na2S at 24 hours after myocardial ischemia and reperfusion may be responsible for inducing miR-21 levels and therefore instigating cyclical activation of Akt and induction of miR-21. 6
In our current study, proteomic analysis by 2D-gel revealed 46 protein spots with altered expression profiles between saline and Na2S-treated mice at 28 days post-MI. Besides cofilin-2, other notable proteins showing increased expression are myosin light chain 3 (MYL3) and VEGF. Myosin light chain 3 modulates the myosin cross-bridge kinetics, and in failing hearts, its cleavage is associated with reduction in myocyte contractile performance. 21 Vascular endothelial growth factor plays a key role in myocardial remodeling as it is involved in new vessel formation, endothelial cell migration and activation, and tissue regeneration. 22 Interestingly, VEGF levels are found to be depleted in patients with heart failure. 23 Hydrogen sulfide is known to regulate VEGF-induced angiogenesis. 24 Further investigation is needed to elucidate the role of VEGF and MYL3 in ischemic HFrEF in the context of H2S donor therapy.
The MALDI-TOF/TOF tandem mass spectrometry identified a meaningful change in cofilin-2 expression with ischemia, which was attenuated with Na2S treatment. Confirmatory Western blot analysis also showed a significant increase in cofilin-2 at 28 days after permanent LAD occlusion that was suppressed with Na2S treatment. Cofilin-2 belongs to the family of actin-binding proteins, and humans express 3 forms including: actin-binding protein (ADF); cofilin-1, the major form in non-muscle tissue; and cofilin-2, the major form in differentiated muscle including cardiac muscle. 25 Cofilin and related ADF family proteins are phosphorylated at a regulatory site Ser3. 25 In its phosphorylated form, cofilin is unable to bind actin, whereas dephosphorylation reactivates its actin-depolymerizing function thus indicating the Ser3 site as an activating switch for cofilin activity. 25 The mechanism by which Na2S regulates cofilin-2 remains unclear in our study. However, because cofilin-2 is a target of miR-21, 26 it is plausible that H2S can act through miR-21-mediated pathway to suppress cofilin-2 expression, although this pathway requires future investigation.
Cofilin-2 expression was sequestered in human tissue samples of idiopathic dilated cardiomyopathy and was associated with an imbalanced ratio of phosphorylated to total cofilin-2 levels. 9 Further studies in neuronal cells treated with N-methyl-D-aspartate (NMDA) demonstrated that cofilin activation mediates Bax translocation to the mitochondria correlating with a loss of transmembrane potential that precedes cytochrome c release and caspase activation. 27 An earlier study in cultured neonatal rat cardiomyocytes showed that NMDA induced alterations causing Bax levels to significantly increase and expression of Bcl-2 to decrease in comparison to cardiomyocytes treated with NMDA-R antagonist. 28 Recent investigations have demonstrated that cofilin-2 and Bax form a protein complex in cardiomyocytes in response to oxidative stress; hence, suggesting that cofilin-2 might function as a Bax transporter to the mitochondria. 29 These studies are in agreement with our results showing significant increase in Bax expression and reduction in Bcl-2/Bax ratio at 28 days post-MI in our saline-treated mice. Conversely, treatment with Na2S reversed these changes.
Only dephosphorylated cofilin translocates to the mitochondria leading to cytochrome c release, whereas the phosphorylated form suppresses mitochondrial translocation and apoptosis. 10 This leads to a significant accumulation of cytochrome c in the cardiomyocyte cytoplasm inducing the apoptotic cascade and leading to progressive decline in left ventricular function. 30 In addition, there is interest in exploring the relationship between cofilin and NLRP3 inflammasome, a complex of nucleotide binding oligomerization domain-like receptor, ASC, and pro-caspase-1. 6, 16 Inflammasome activation, proposed to be mediated via cation movement, mitochondrial membrane dysfunction, and production of reactive oxygen species, can induce pyroptosis leading to reduction of cardiac contractility and HF progression. 31 A recent study demonstrated the inhibitory effects of H2S on NLRP3 inflammasome activation both in vitro and in vivo, strongly suggesting a potential therapeutic role for H2S in inflammation resolution. 32 Further studies are warranted to examine the involvement of cofilin-2 in this pathway. A potential limitation of this study is the focus on male mice, which was chosen to avoid potential confounding sex-related factors, if any. Future studies are needed to test the role of cofilin-2 in female mice subjected to MI as well as the impact of H2S donors.
Conclusions
In summary, we showed that chronic Na2S treatment mitigates LV remodeling in MI-induced HF through reducing fibrosis, inflammasome activation, and apoptotic signaling. For the first time, we showed that Na2S modulates the expression of cofilin-2. Therefore, with the continuous development of novel H2S donors, many of which are in the pipeline, and based on the recently published phase I clinical study on SG1002 demonstrating its safety and tolerability in HF patients as well as its ability to reduce BNP levels, long-term supplementation of “therapeutic” levels of H2S could be a viable modality in the management and treatment of ischemic cardiomyopathy.
Supplemental Material
Supplemental Material, Supplemental_Figure_1 - Hydrogen Sulfide Therapy Suppresses Cofilin-2 and Attenuates Ischemic Heart Failure in a Mouse Model of Myocardial Infarction
Supplemental Material, Supplemental_Figure_1 for Hydrogen Sulfide Therapy Suppresses Cofilin-2 and Attenuates Ischemic Heart Failure in a Mouse Model of Myocardial Infarction by Khoa Nguyen, Vinh Q. Chau, Adolfo G. Mauro, David Durrant, Stefano Toldo, Antonio Abbate, Anindita Das and Fadi N. Salloum in Journal of Cardiovascular Pharmacology and Therapeutics
Supplemental Material
Supplemental Material, Supplemental_Figure_2 - Hydrogen Sulfide Therapy Suppresses Cofilin-2 and Attenuates Ischemic Heart Failure in a Mouse Model of Myocardial Infarction
Supplemental Material, Supplemental_Figure_2 for Hydrogen Sulfide Therapy Suppresses Cofilin-2 and Attenuates Ischemic Heart Failure in a Mouse Model of Myocardial Infarction by Khoa Nguyen, Vinh Q. Chau, Adolfo G. Mauro, David Durrant, Stefano Toldo, Antonio Abbate, Anindita Das and Fadi N. Salloum in Journal of Cardiovascular Pharmacology and Therapeutics
Supplemental Material
Supplemental Material, Supplement_final - Hydrogen Sulfide Therapy Suppresses Cofilin-2 and Attenuates Ischemic Heart Failure in a Mouse Model of Myocardial Infarction
Supplemental Material, Supplement_final for Hydrogen Sulfide Therapy Suppresses Cofilin-2 and Attenuates Ischemic Heart Failure in a Mouse Model of Myocardial Infarction by Khoa Nguyen, Vinh Q. Chau, Adolfo G. Mauro, David Durrant, Stefano Toldo, Antonio Abbate, Anindita Das and Fadi N. Salloum in Journal of Cardiovascular Pharmacology and Therapeutics
Footnotes
Acknowledgments
The authors would like to thank Jun He, M.S. for her help with surgical preparation and statistical analysis.
Author Contributions
K.N., V.Q.C., A.G.M. and F.N.S. planned the study, executed experiments and analyzed data, and drafted the manuscript. D.D., S.T., A.A., and A.D. executed experiments and interpreted data. F.N.S. approved final manuscript with acknowledgment from all authors.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported in part by grants from the American Heart Association (14GRNT20010003) and the National Institutes of Health (R01HL133167 and R01HL142281) to Dr. Salloum.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.