
Abstract
Background:
Atrial fibrillation (AF) is an important and growing clinical problem. Current pharmacological treatments are unsatisfactory. Electrical remodeling has been identified as one of the principal pathophysiological mechanisms that promote AF, but there are no effective therapies to prevent or correct electrical remodeling in patients with AF. In AF, cardiac production and circulating levels of B-type natriuretic peptide (BNP) are increased. However, its functional significance in AF remains to be determined. We assessed the hypotheses that chronic BNP treatment may prevent the altered electrophysiology in AF, and preventing AF-induced activation of Ca2+/calmodulin-dependent protein kinase II (CaMKII) may play a role.
Methods and Results:
Forty-four rabbits were randomly divided into sham, rapid atrial pacing (RAP at 600 beats/min for 3 weeks), RAP/BNP, and sham/BNP groups. Rabbits in the RAP/BNP and sham/BNP groups received subcutaneous BNP (20 μg/kg twice daily) during the 3-week study period. HL-1 cells were subjected to rapid field stimulation for 24 hours in the presence or absence of BNP, KN-93 (a CaMKII inhibitor), or KN-92 (a nonactive analog of KN-93). We compared atrial electrical remodeling-related alterations in the ion channel/function/expression of these animals. We found that only in the RAP group, AF inducibility was significantly increased, atrial effective refractory periods and action potential duration were reduced, and the density of I Ca, L and I to decreased, while I K1 increased. The changes in the expressions of Cav1.2, Kv4.3, and Kir2.1 and currents showed a similar trend. In addition, in the RAP group, the activation of CaMKIIδ and phosphorylation of ryanodine receptor 2 and phospholamban significantly increased. Importantly, these changes were prevented in the RAP/BNP group, which were further validated by in vitro studies.
Conclusions:
Chronic BNP therapy prevents atrial electrical remodeling in AF. Inhibition of CaMKII activation plays an important role to its anti-AF efficacy in this model.
Introduction
Atrial fibrillation (AF) is the most commonly sustained arrhythmia that is associated with pronounced morbidity, mortality, and socioeconomic burden. Over the years, although many important advances in our understanding and treatment of AF have been made, it remains a serious and growing health problem. Presently available therapeutic approaches have limited efficacy and potential adverse effects. 1 -5
Now it is increasingly recognized that the basic arrhythmia mechanisms of AF are rapid focal ectopic firing and/or reentry. Accumulating evidence has shown that electrical remodeling promotes both for the induction of triggered ectopic activity and for the activation of Ca2+-related cell signaling that mediates profibrillatory remodeling. Electrical remodeling entails atrial electrophysiological property changes, including the alterations that occur in ion channels, pumps, and exchangers in atrial cells. 3,5 -7 Ca2+/calmodulin-dependent protein kinase II (CaMKII) has been identified as a critical player in promoting these electrophysiological changes. Ca2+/calmodulin-dependent protein kinase II modulates several of the pathways involved in AF and atrial electrical remodeling. However, there are currently no CaMKII inhibitors available clinically. There is an urgent need for effective therapies to prevent and to correct electrical remodeling in AF. 5,8 Thus, it may be the best to use some complementary approaches to CaMKII inhibition.
Emerging evidence supports B-type natriuretic peptide (BNP) as a new therapeutic option for the treatment in AF. In AF, cardiac production and circulating levels of BNP are increased. It is well known that BNP produced by both atrial myocytes and ventricular myocytes exhibits a range of actions. In addition to its vasodilating and natriuresis actions, BNP has other cardiac protective effects, such as antifibrotic, antihypertrophic, and renin–angiotensin–aldosterone inhibition. 9 -12 These properties are beneficial in AF. Recombinant human BNP (rhBNP) has been recommended to treat acute heart failure (HF). 13,14 Furthermore, rhBNP therapy has been shown to reduce the new onset of AF in HF patients. 12,15,16 Although the beneficial actions have been well-documented in HF patients, its functional effect in AF is unclear. No studies have been reported about the response after chronic BNP therapy in AF.
With this in mind, we assessed the hypothesis that chronic BNP therapy may prevent atrial electrical remodeling in AF. We extended the hypothesis to include the demonstration of the prevention of AF-associated increased activation of CaMKII with chronic BNP treatment. Accordingly, we examined the antiarrhythmic efficacy and underlying electrophysiological mechanisms of chronic BNP therapy in a well-established rabbit model of AF induced by rapid atrial pacing (RAP).
Materials and Methods
Rapid Atrial Pacing Animal Model
This investigation was approved by the Animal Experimentation Ethics Committee of Harbin Medical University (HMU) and conformed to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (no. 85-23).
Healthy adult New Zealand white rabbits (male, 2.5-3.0 kg, aged 4-5 months) were obtained from the Experimental Animal Center of HMU (China). The rabbit model of AF was established as reported previously. 17,18 In the current study, the surface electrocardiogram was checked for every rabbit. The rabbits were in normal sinus rhythm and had no detectable arrhythmia before therapy. The rabbits were anesthetized with ketamine (35 mg/kg) and xylazine (5 mg/kg; Sigma Aldrich, St Louis, Missouri). A pacemaker (Harbin Polytechnic University, Harbin, China) was inserted into a subcutaneous (SC) pocket on the back of the rabbit. The pacemaker electrode was sutured to the right atrium. The animals were confined for 1 week after the surgery for recovery.
The rabbits were randomized into 4 groups (n = 11 per group, 3 for the isolation of left atrial myocytes and whole-cell patch-clamp recording and 8 for other analyses). (1) Sham group: sham surgery and no pacing; (2) RAP group: RAP at 600 beats/min for 3 weeks; (3) RAP/BNP group: RAP at 600 beats/min and SC administration of rhBNP (Nuodikang Biological Pharmaceutical Company Ltd, Chengdu, China) at a dose of 20 μg/kg twice daily for 3 weeks; and (4) sham/BNP group: sham surgery and SC rhBNP therapy (20 μg/kg, twice daily) for 3 weeks. The SC route of administration of rhBNP was used as reported previously. The rhBNP dose regimen was based on previous studies and our initial dosage testing, which had minimal effect on systemic blood pressure and heart rate of the rabbits. The dosage used in the current study was in the range of therapeutic doses, but slightly lower than that used in the past for the treatment of systolic HF patients. 19 -22
HL-1 Cell Model
To determine that the prevention of AF-induced activation of CaMKII may play a critical role to the antielectrical remodeling by BNP, in the current study, we utilized a well-established HL-1 cell model of AF 17,23 -27 and applied CaMKII inhibitor (KN-93) in this model. This approach, to use KN-93 in the HL-1 cell model of AF, will allow a better comparison and minimizing the off-target effects of KN-93. It has been well-documented that HL-1 cells are derived from mouse atrial tumor (AT-1) cells and can proliferate indefinitely in culture. They retain a highly differentiated cardiac morphological, biochemical, and electrophysiological phenotype reflective of atrial cardiomyocytes. 24,25 Rapid stimulation of HL-1 cells produces electrical remodeling, recapitulating principal phenotypic features of atrial tachycardia remodeling in vivo. HL-1 cells express CaMKIIδ, RyR2, and PLN. 28,29 Ca2+/calmodulin-dependent protein kinase II is a serine/threonine protein kinase expressed in many cell types. Given the ubiquitous nature of CaMKII and its multifunctional role in several physiological processes, to avoid the off-target effects of KN-93 by systemic use on the rabbits, we studied the HL-1 cells to further validate the role of CaMKII. 5,30
HL-1 cells were subjected to rapid field stimulation (10 Hz, 5 milliseconds) for 24 hours using a YC-2 stimulator (Chengdu, China). The HL-1 cells were divided into 6 groups: (1) control group: cultured for 24 hours, no rapid field stimulation or other drug interventions; (2) BNP group: cultured for 24 hours with mouse BNP (100 nM, GL Biochem, Shanghai, China) and no rapid field stimulation; (3) AF group: rapid field stimulation for 24 hours as aforementioned; (4) AF/KN-93 group: rapid field stimulation for 24 hours with KN-93 (a CaMKII inhibitor, 1 μM; APExBIO, Houston, Texas) pretreatment 1 hour; (5) AF/KN-92 group: rapid field stimulation for 24 hours with KN-92 (a nonactive analog of KN-93, 1 μM; APExBIO, Houston, Texas) pretreatment for 1 hour; and (6) AF/BNP group: rapid field stimulation for 24 hours with mouse BNP (100 nM; GL Biochem, Shanghai, China) pretreatment for 1 hour.
Determine AF Induction Rate and Atrial Effective Refractory Period
The rabbit atrial electrophysiology study was performed as described previously. 17,18 In these animals, atrial effective refractory periods (AERPs) were measured at 2 basic cycle lengths (150 and 200 milliseconds), which were recorded as AERP150 ms and AERP200 ms, respectively. After the AERPs were measured, AF was induced by applying 10 seconds burst pacing (10 Hz, 2 milliseconds) to the right atrium at 4 times the threshold current. Each rabbit was induced 10 times. Atrial fibrillation induction rate in each group was calculated as (AF times)/(induction times, ie, 80). 31
Determine N-Terminal Pro-BNP Levels
Blood collections were made before the surgery at the start of the study and after 3 weeks of RAP at the end of the study. Serum N-terminal pro-BNP (NT-proBNP) level was determined by an enzyme-linked immunosorbent assay kit (Shanghai Jianglai Biotech, Shanghai, China) following the manufacturer’s instructions.
N-terminal pro-BNP is the amino terminal cleavage fragment of endogenous BNP and is secreted in equimolar amounts with BNP. Therefore, NT-proBNP concentration can be applied to denote the release of endogenous BNP. We measured serum NT-proBNP in this study because it is not affected by exogenous rhBNP and has higher stability and longer half-life. 32
Cell Isolation
Left atrial myocytes were isolated from the hearts of the rabbits as described previously. 33 After the rabbits were anesthetized, their hearts were quickly excised and mounted on a Langendorff apparatus. Then the hearts were perfused with the following solutions in sequence: buffer A (75 mL Ca2+-free Tyrode), buffer B (40 mL Ca2+-free Tyrode, 10 µL 200 mM Ca2+, 400 µL 100 mg/mL bovine serum albumin [BSA]), buffer C (40 mL Ca2+-free Tyrode, 200 µL 200 mM Ca2+, 400 µL 100 mg/mL BSA), buffer D (50 mL Ca2+-free Tyrode, 12.5 µL 200 mM Ca2+, 500 µL 100 mg/mL BSA), buffer E (35 mL buffer D, 31.5 mg collagenase), and buffer F (15 mL buffer D, 13.5 mg collagenase). Then, the left atria were separated from the hearts. The myocardia of left atria were minced and filtered. The freshly isolated left atrial myocytes were gently centrifuged and resuspended in the storage solution for subsequent patch clamp experiments.
Whole-Cell Patch Clamp Recording
Myocyte ion channel responses were recorded with a patch clamp amplifier (HEKA EPC10, Ludwigshafen, Germany). Action potential duration (APD) was recorded at 37°C under current clamp mode. Current densities (pA/pF) were obtained by current amplitudes normalized to cell capacitance in each cell (n = 9 cells from 3 rabbits per group). The amplitude of L-type calcium current (I Ca, L), transient outward potassium current (I to), and inward rectifier potassium current (I K1) were measured as described previously. 33
The pipette solution for I Ca, L contained (mM) 1 MgCl2, 120 CsCl, 10 EGTA, 10 HEPES, 5 phosphocreatine disodium salt, 4 Mg-ATP, and 0.3 Na2-GTP (pH adjusted to 7.20 with CsOH). The pipette solution for potassium current (I to and I K1) and action potential (AP) recordings contained (mM) 0.1 CaCl2, 140 K-gluconate, 1 MgCl2, 5 NaCl, 10 HEPES, 2 Mg-ATP, and 1 EGTA (pH adjusted to 7.20 with KOH). The extracellular solution for I Ca, L recordings contained (mM) 140 TEA-Cl, 10 HEPES, 2 MgCl2, 10 CaCl2, and 5 glucose (pH adjusted to 7.4 with TEA-OH). The extracellular solution for potassium currents and AP recording contained (mM) 3.5 KCl, 2 mM CaCl2,140 NaCl, 1 MgCl2, 10 glucose, 1.25 NaH2PO4, and 10 HEPES (pH adjusted to 7.4 with NaOH). To record I K1 and I to, both I Ca, L and I Na were blocked with 0.2 mM CdCl2 and 30 μM TTX, respectively.
Real-Time Polymerase Chain Reaction
From the left atria of the rabbit, total RNA was extracted using TRIzol reagent (BioTeke, Beijing, China). Using M-MLV reverse transcriptase (BioTeke), total RNA was reverse-transcribed to obtain complementary DNA. The messenger RNA (mRNA) levels of rabbit BNP, Cav1.2 (the subunit of L-type calcium channel), Kv4.3 (the subunit of transient outward potassium channel), and Kir2.1 (the subunit of inward rectifier potassium channel) were determined using SYBR Green I incorporation method on an ABI 7500 fast real-time polymerase chain reaction system (Applied Biosystems, Foster, California). β-Actin was used as an internal control. The primers of related genes used in this study are listed in Table 1.
Primers for Real-Time PCR.

Abbreviations: BNP, B-type natriuretic peptide; PCR, polymerase chain reaction.
Western Blot
Protein samples were extracted from the left atria of the rabbits or from HL-1 cells. Western blot analysis was performed as described previously. 34 Primary antibodies included p-CaMKIIδ antibody (1:500; Santa Cruz, Dallas, Texas), anti-oxidized-CaMKII (Met281/282) antibody (ox-CaMKIIδ, 1:1000; Merck, Temecula, California), RYR2 (pSer2814) pAb (1:5000; Badrilla, Leeds, UK), and phospho-phospholamban (Thr17) antibody (1:2500; Thermo, Waltham, Massachusetts). Secondary antibody was horseradish peroxidase-conjugated goat anti-rabbit or rabbit anti-mouse immunoglobulin G (1:1000; Zhongshan, Beijing, China). Western blot bands were normalized to β-actin as an internal control.
Statistical Analyses
SPSS 19.0 software was used for statistical analyses. All data are presented as mean ± standard error of the mean, except for AF induction rate as a percentage. Statistical significance between different groups was determined by 1-way analysis of variance (ANOVA) and least significant difference post hoc test. Serum concentrations of NT-proBNP in 4 groups before and after 3 weeks of RAP were compared with 2-factor ANOVA. The AF induction rate was compared with χ2 test. A value of P < .05 was considered statistically significant.
Results
Effects of Chronic rhBNP Therapy on AF Inducibility and AERPs After 3 Weeks of RAP
As displayed in Figure 1, the AF induction rate increased significantly in the RAP group compared with the sham group (RAP: 51.3% vs sham: 7.5%). Both AERP200 ms and AERP150 ms were significantly shortened (AERP200 ms:78.3 ± 9.2 milliseconds vs 100.3 ± 16.3 milliseconds; AERP150 ms: 75.3 ± 9.3 milliseconds vs 94.8 ± 15.6 milliseconds). Importantly, in the RAP/BNP group, the AF induction rate significantly decreased to 13.8%, the AERP200 ms and AERP150 ms reached to 95.8 ± 16.7 milliseconds and 92.0 ± 18.1 milliseconds, respectively. There was no significant difference in AF induction rate, AERP200 ms, or AERP150 ms between the RAP/BNP and sham groups (Figure 1A-C).

Group means data (n = 8/group) of the effects of rapid atrial pacing (RAP) and chronic recombinant human BNP (rhBNP) therapy on AF induction rate (A), AERP200 ms (B) and AERP150 ms (C), and mRNA expression of BNP (D) in left atrial myocytes. Compared with same animals, in the untreated RAP group, the AF induction rate, AERP, and mRNA of BNP in the left atrial myocytes were significantly increased. In contrast, in the rhBNP-treated RAP group, these changes in left atrial myocytes were prevented. *P < .05 versus sham group; # P < .05, RAP/BNP versus RAP group. AERP indicates atrial effective refractory period; AF, atrial fibrillation; BNP, B-type natriuretic peptide; mRNA, messenger RNA.
Effects of Chronic rhBNP Therapy on APD, I Ca, L, I to, and I K1 of the Left Atrial Myocytes After 3 Weeks of RAP
Figure 2 presents the effects of chronic rhBNP therapy on the APD of the left atrial myocytes obtained from the RAP rabbits. After 3 weeks of RAP, both APD90 and APD50 were shortened significantly compared with the sham group. In the RAP/BNP group, these changes were nearly prevented. Figure 3 exhibits the effects of chronic rhBNP therapy on I Ca, L and the expression of Cav1.2 in the left atrial myocytes obtained from the RAP rabbits. After 3 weeks of RAP, the current density of I Ca, L and the expressions of Cav1.2 were significantly decreased. Figures 4 and 5 show the effects of chronic rhBNP therapy on I to, I K1, and the expressions of Kv4.3 and Kir2.1 in the left atrial myocytes obtained from the RAP rabbits. As shown in Figure 4, the current density of I to significantly decreased and Kv4.3 significantly reduced after 3 weeks of RAP. In contrast, as demonstrated in Figure 5, both the current density of I K1 and the expressions of Kir2.1 significantly increased after 3 weeks of RAP. Importantly, these changes were inhibited significantly after chronic rhBNP therapy.
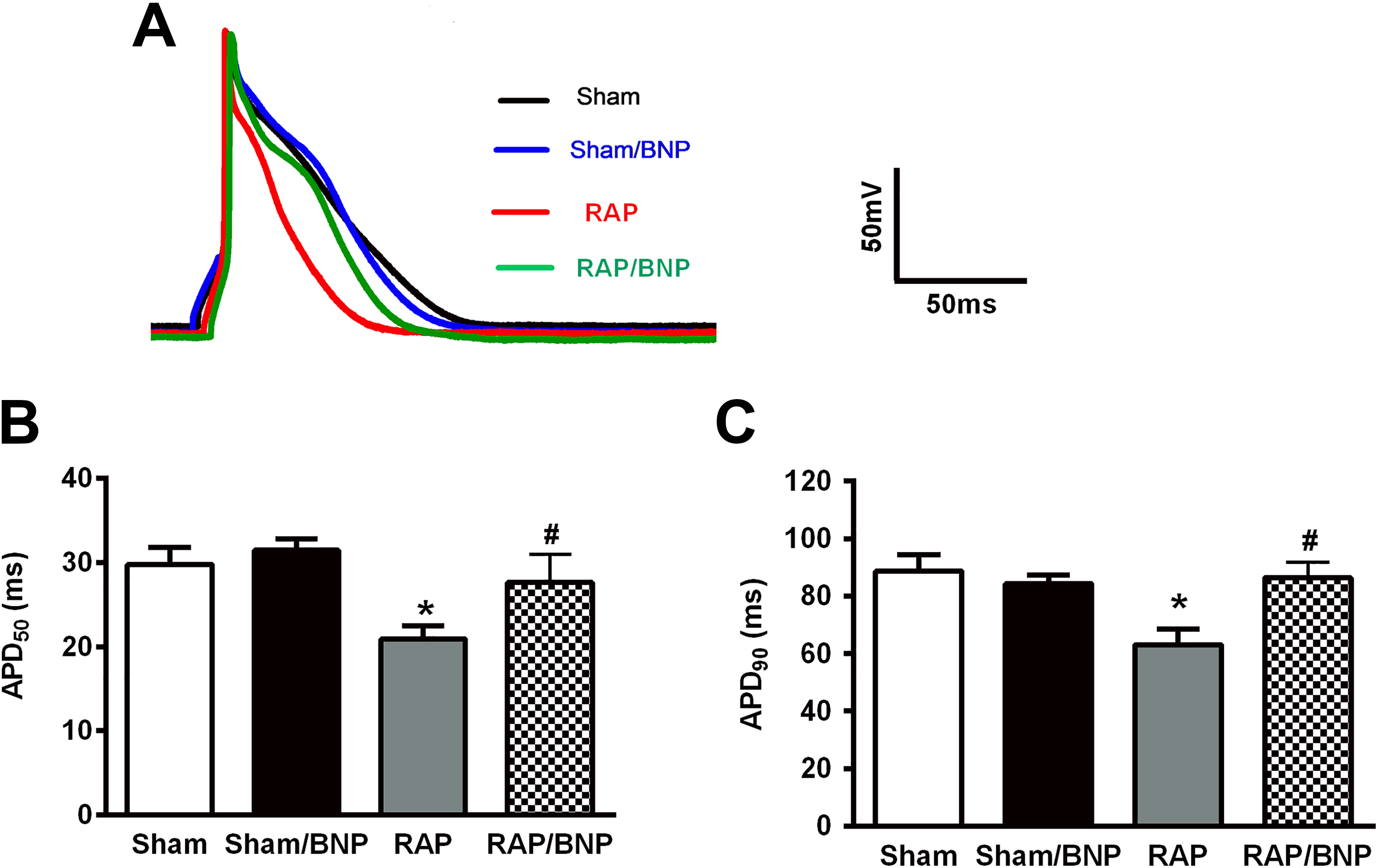
The effects of rapid atrial pacing (RAP) and chronic recombinant human BNP (rhBNP) therapy on the action potential duration (APD) of the left atrial myocytes. (A) Examples of the traces of APD of single atrial myocytes by patch clamp technique; (B and C) group means data of APD50 and APD90 of single atrial myocytes, respectively (n = 9 cells from 3 rabbits per group). It is noted, only in the untreated RAP group, both APD50 and APD90 in the left atrial myocytes were significantly shortened. *P < .05 versus sham group; # P < .05, RAP/BNP versus RAP group. BNP indicates B-type natriuretic peptide.

The effects of rapid atrial pacing (RAP) and chronic recombinant human BNP (rhBNP) therapy on the L-type calcium current (I Ca, L) and the expression of Cav1.2 in the left atrial myocytes. (A) Group means of the I Ca, L density (n = 9 cells from 3 rabbits per group); (B) group means of the expression of Cav1.2 (n = 6/group). *P < .05 versus sham group; # P < .05, RAP/BNP versus RAP group. BNP indicates B-type natriuretic peptide.

The effects of rapid atrial pacing (RAP) and chronic recombinant human BNP (rhBNP) therapy on the transient outward potassium current (I to) and the expression of Kv4.3 in the left atrial myocytes. (A) Group means of I to density (n = 9 cells from 3 rabbits per group); (B) group means of the expression of Kv4.3 (n = 6/group). *P < .05, versus sham group; # P < .05, RAP/BNP versus RAP group. BNP indicates B-type natriuretic peptide.
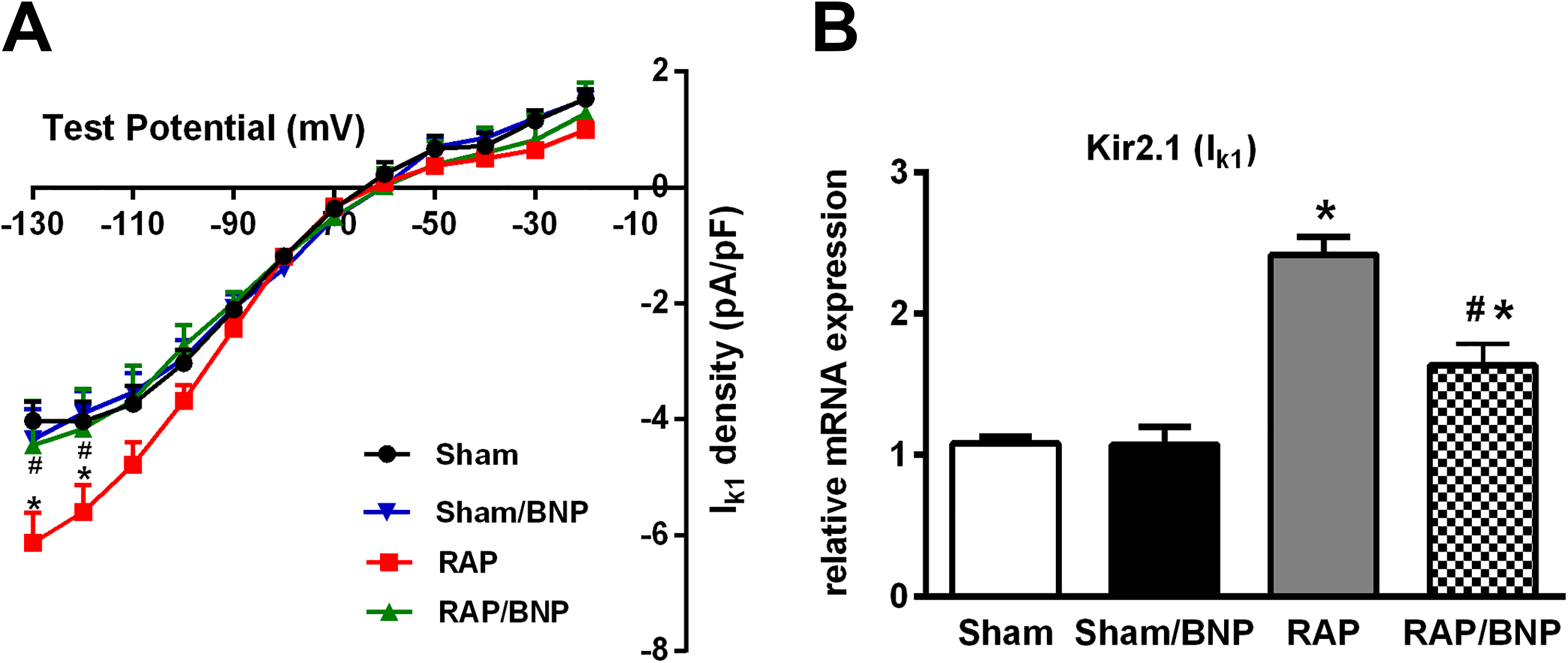
The effects of rapid atrial pacing (RAP) and chronic recombinant human BNP (rhBNP) therapy on the inward rectifier potassium current (I K1) and the expression of Kir2.1 in the left atrial myocytes. (A) Group means of I K1 density (n = 9 cells from 3 rabbits per group); (B) group means of the expression of Kir2.1 (n = 6/group). *P < .05 versus sham group; # P < .05, RAP/BNP versus RAP group. BNP indicates B-type natriuretic peptide.
Effects of Chronic rhBNP Therapy on the mRNA Expression and Plasma Levels of BNP After 3 Weeks of RAP
As exhibited in Figure 1D and summarized in Table 2, the mRNA expression of BNP increased significantly in the RAP group. The increase was attenuated significantly in the RAP/BNP group. There was no significant difference in NT-proBNP concentration among 4 groups before surgery. However, after 3 weeks of RAP, NT-proBNP concentration increased significantly in the RAP group compared with that in the sham animals or preoperation. The increase was inhibited significantly in the RAP/BNP group.
Serum Concentration of NT-proBNP in 4 Groups Before and After 3 Weeks of Rapid Atrial Pacing (RAP).a

Abbreviations: BNP, B-type natriuretic peptide; NT-proBNP, N-terminal pro-BNP; RAP, rapid atrial pacing; SEM, standard error of the mean.
aValues are presented as pg/mL and as mean ± SEM; n = 8 per group.
b P < .05 versus baseline.
c P < .05 versus sham group.
d P < .05, RAP versus RAP/BNP group.
Effects of Chronic BNP Therapy on Activation of CaMKII in the Rabbit and HL-1 Cells Model of AF
Figure 6 displays the effects of chronic rhBNP therapy on the activation of CaMKII signaling pathway in the left atrial myocytes obtained from the RAP rabbits. As demonstrated, the RAP group showed increased phosphorylation and oxidation of CaMKIIδ, phosphorylation of RyR2, and PLN compared with the sham group, while BNP treatment attenuated this increase. These results were further verified by the findings from HL-1 cells. As exhibited in Figure 7, in the AF group, HL-1 cells, p-CaMKIIδ, ox-CaMKIIδ, p-RyR2, and p-PLN increased significantly compared with the control group. KN-93 is a CaMKII inhibitor, which decreases CaMKII-mediated phosphorylation of RyR2 and PLN under stress conditions. B-type natriuretic peptide effectively attenuated the upregulation of p-CaMKIIδ, ox-CaMKIIδ, p-RyR2, and p-PLN in HL-1 cells under rapid field stimulation similar to KN-93, while KN-92 treatment did not show any obvious effects.

The effects of rapid atrial pacing (RAP) and chronic recombinant human BNP (rhBNP) therapy on the activation of CaMKII signaling pathway in the left atrial myocytes. (A) Representative bands and p-CaMKIIδ expression (phosphorylation of CaMKIIδ at Thr287) obtained from 4 group rabbits; (B) representative bands and quantitative analyses of ox-CaMKIIδ (oxidation of CaMKIIδ at Met281/282); (C) representative bands and quantitative analyses of p-PLN (phosphorylation of PLN at Thr17); (D) representative bands and quantitative analyses of p-RyR2 (phosphorylation of at Ser2814). *P < .05 versus sham group; # P < .05, RAP/BNP versus RAP group, n = 6/group. BNP indicates B-type natriuretic peptide; CaMKII, Ca2+/calmodulin-dependent protein kinase II.

The exogenous BNP administration on the activation of CaMKII signaling pathway in the HL-1 cells model of AF. HL-1 cells were subjected to rapid field stimulation for 24 hours in the presence or absence of BNP, KN-93 (a CaMKII inhibitor), or KN-92 (a nonactive analog of KN-93). (A) Representative bands and quantitative analyses of p-CaMKIIδ (phosphorylation of CaMKIIδ at Thr287); (B) representative bands and quantitative analyses of ox-CaMKIIδ (oxidation of CaMKIIδ at Met281/282); (C) representative bands and quantitative analyses of p-PLN (phosphorylation of PLN at Thr17); (D) representative bands and quantitative analyses of p-RyR2 (phosphorylation of RyR2 at Ser2814). *P < .05 versus control (ctrl) group; # P < .05 versus AF group, n = 5/group. AF indicates atrial fibrillation; BNP, B-type natriuretic peptide; CaMKII, Ca2+/calmodulin-dependent protein kinase II.
Discussion
In the present study, we showed, for the first time, chronic rhBNP therapy in a rabbit model of AF results in the prevention of atrial electrical remodeling, CaMKII activation, and increased expression and secretion of endogenous BNP. The prevalence and burden of AF worldwide call for an urgent increase in investment in basic and translational research. 1 -3 The current investigation provides evidence supporting BNP as an effective therapeutic option for the treatment in AF. This study has significant translational value and high public health importance.
Atrial Electrical Remodeling, CaMKII Activation, and Chronic BNP Therapy
Previously, few clinical studied in patients with AF reported antiarrhythmic efficacy after BNP administration. Intravenous administration of rhBNP in patients with acute HF reduced the new onset of AF. Earlier studies showed that perioperative nesiritide (rhBNP) or carperitide (recombinant human ANP) infusion reduced the occurrence of postoperative AF in patients undergoing coronary artery bypass grafting during hospital stay. In another study, nesiritide therapy caused a significantly reduced new-onset AF in patients with acute decompensated HF during a 90-day follow-up period. 12,15,16 However, no previous AF studies have assessed the effects after BNP administration on the alterations of electrical remodeling.
Conceptually, AF induction requires a vulnerable substrate and a trigger that acts on the substrate to initiate the arrhythmia. It is increasingly recognized that principal electrophysiological mechanisms contributing to AF include electrical remodeling, structural remodeling, autonomic remodeling, and Ca2+-handling abnormalities. Abbreviations of refractories mainly due to reduction in APD provide substrate for reentry. Increases in delayed afterpolarization (DAD) activity due to Ca2+-handling abnormalities promote local ectopic firing Ca2+-handling abnormalities. 3,5,35,36 In the current investigation, consistent with previous experimental and human studies, the untreated RAP animals showed characteristics typical of altered electrophysiology, including in the current density and in ion channel mRNA or protein expressions. We found that in the left atrial myocytes isolated from the untreated RAP animals, atrial electrical remodeling was characterized mainly by the significantly shortening of APD and AERP and the changes of the atrial ion current, which were predominantly mediated via the changes in the expression of the ion channels. The shortening of APD/AERP would be expected to reduce the excitation wavelength given by conduction velocity (CV) × AERP. The additional substrate would be abnormal Ca2+ handling, which could generate DADs (and therefore triggered activity) and contribute to reentrant substrates. 35,36
Decreased I Ca, L is the principal component of electrical remodeling, which is mainly due to the reduced expression of the subunit of I Ca, L (Cav1.2) to prevent potentially cytotoxic Ca2+ overload. Both the reduced I Ca, L and the increased I K1 may account for the reduced APD. The increased expression of Kir2.1 subunit results in I K1 increase, while the downregulation of the Kv4.3 subunit causes a decrease in I to. 7,30,37 -39 Importantly, we found that in the left atrial myocytes isolated from the RAP/BNP animals, the AF-related APD shortening and the loss of AERP with associated characteristical changes on the downregulation of I Ca, L, I to and the upregulation of I K1, as well as the altered expression of atrial ion channels genes (Cav1.2, Kv4.3, and Kir2.1), were all absent. There were no significances in these measurements between sham and RAP/BNP animals, thus demonstrating atrial electrical remodeling was prevented by chronic BNP therapy.
How does chronic BNP administration prevent AF-induced electrical remodeling? In the current investigation, the prevention of AF-induced activation of CaMKII might play a critical role in the antielectrical remodeling by chronic BNP.
Excessive CaMKII activation has been implicated in AF triggering and reentry mechanisms through its downstream effects on ion channels and proarrhythmic remodeling that results from cell death, fibrosis, and hypertrophy. It has been well-documented that CaMKIIδ activation can be promoted by atrial tachycardia, oxidative stress, and sympathetic hyperactivity and results in phosphorylation of ion channels and Ca2+-cycling proteins, including I Ca, L, RyR2, and PLN, thereby facilitating cell membrane hyperexcitability and afterdepolarizations and promotes the development of AF and atrial electrical remodeling. 4,5,30,40 Previous studies on total CaMKIIδ protein expression in AF were not conclusive; some studies reported an increase in total CaMKIIδ protein expression in AF, while others reported no change in expression. 5,40,41 In the current study, we analyzed the phosphorylation and oxidation of CaMKIIδ instead of total CaMKIIδ protein expression. We observed that in untreated RAP animals, RAP for 3 weeks promoted autophosphorylation and oxidation of CaMKIIδ with the phosphorylation of RyR2 (phosphorylation of Ser2814) and PLN (phosphorylation of Thr17). Similar results were obtained from the in vitro HL-1 cell model of AF. These findings are consistent with previous reports and demonstrate that CaMKIIδ activation is an important molecular mechanism of atrial electrical remodeling in AF. Of importance, we further found that these alterations were prevented by a 3-week rhBNP therapy in the RAP/BNP animals. We further validated the effects of exogenous BNP on the activation of CaMKII signaling pathway in HL-1 cells. Previous reports showed that rapid stimulation of HL-1 cells produced electrical remodeling, recapitulating principal phenotypic features of atrial tachycardia remodeling in vivo. 23 -26 Ca2+/calmodulin-dependent protein kinase II is a nodal proarrhythmic signal. The changes of p-CaMKII, ox-CaMKII, p-PLN, and pRyR2 in HL-1 cells were similar to that of the rabbits. It is noted that KN-93 exerted similar effects to BNP in terms of expression of p-CaMKII, ox-CaMKII, p-PLN, and pRyR2.
Many previous studies have demonstrated that inhibiting CaMKII activation and/or elimination of CaMKII-dependent RyR2/PLN phosphorylation is an important molecular mechanism that protects animals from developing AF. Also, in fibrillating human atrial cells, CaMKII inhibition appears to be able to correct proarrhythmic, defective intracellular Ca2 + homeostasis. However, there are currently no CaMKII inhibitors available clinically. 5,30,42,43
B-Type Natriuretic Peptide Activation and Chronic BNP Therapy
Both BNP and NT-proBNP are applied as diagnosis, management, and prognostic tool for HF. It is now also clear that elevated BNP levels are closely associated with AF. Plasma BNP levels are increased in patients with paroxysmal, persistent and permanent AF, and normal left ventricle function. It has been shown that both BNP and NT-proBNP are significantly elevated in patients with lone AF. Both BNP and NT-proBNP levels decrease significantly after catheter ablation of AF. 9,10,44,45 In agreement with the clinical observations, in the current study, we found that the mRNA expression and the release of endogenous BNP were significantly increased in the untreated RAP rabbits. Moreover, we further found that the elevated BNP and NT-proBNP were significantly reduced and nearly restored to the levels in the sham rabbits. Our current findings not only support the diagnostic and prognostic value of BNP as a biomarker in AF but also provide new evidence that BNP is an effective therapeutic choice in the management of AF.
It should be realized that BNP has properties that are beneficial in AF and are important for the prevention of CaMKII activation. It is well known that BNP is released from both atrial myocytes and ventricular myocytes. In addition to its effects of diuresis, natriuresis, vasodilation, antihypertrophy, and antifibrosis, BNP has been found to suppress sympathetic tone and the renin–angiotensin system, inhibiting endothelin and vasopressin and imporving cardiac β-adrenergic regulation. It is reported that BNP can act in a paracrine and endocrine manner in many cardiovascular diseases. 9,11,12,46 Although little is known about the electrophysiological effects of BNP, it has been speculated that BNP may have similar properties to ANP, which has been shown to affect the sodium, calcium, and potassium channels and prevent the shortening of the AERP and monophasic APD in a canine RAP model. 47 Importantly, cardiac production and plasma levels of BNP are increased in AF, which may alter left ventricular and atrial functional responses to BNP and suggesting that it is released as a cytoprotective mechanism. However, the changes of BNP-induced cardiac response in AF are not clear. Moreover, no previous studies have examined antiarrhythmic efficacy after BNP therapy in AF. Further studies are needed to improve the understanding of their therapeutic values.
Study Limitations
First, our observations were obtained from an RAP rabbit model. Although this RAP model mimics “lone AF,” it cannot fully represent the complex clinical spectrum of human AF. Second, in the current investigation, we focused on study that systemically assessed electrical remodeling with and without chronic BNP therapy in AF model. We showed that BNP to protect against AF was due to its effects on the prevention and/or correction of electrical remodeling in fibrillating atrial myocytes. However, it is possible that the improvements in AF-related structure remodeling and intracellular calcium-handling remodeling by BNP might also contribute to our current observations. Histology examination was not performed in the current study. The absence of histology is another limitation. Third, altered intracellular Ca2+ dynamics are characteristically observed in cardiomyocytes from AF. The current study did not determine [Ca2+]i transients. More insights will be gained by confocal microscopy assessments. It should be assessed in the future.
Conclusions
Chronic BNP therapy prevents atrial electrical remodeling in a rabbit model of AF induced by RAP. The inhibition of the activation of CaMKII may have contributed to the anti-atrial electrical remodeling effects of BNP. It suggests that chronic BNP may be a useful therapeutic choice for AF.
Footnotes
Author Contributions
Hongyan Zhao contributed to acquisition, analysis and interpretation, and drafted the manuscript. Tiankai Li contributed to analysis and drafted the manuscript. Guangzhong Liu, Li Zhang, Guangnan Li, Jia Yu, Qi Lou, Rui He, Chengchuang Zhan, Luyifei Li, Wen Yang, and Yanxiang Zang contributed to acquisition. Cheping Cheng and Weimin Li contributed to conception and design, and drafted the manuscript. All authors critically revised article, gave final approval. All authors agree to be accountable for all aspects of work ensuring integrity and accuracy.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by grants from Fundamental Research Funds for the Provincial Universities (2017LCZX08), Heilongjiang Postdoctoral Fund (LBH-Z17226), the Health Commission of Heilongjiang Province (2014-376), the National Institutes of Health grant (R01AG049770), and National Natural Science Foundation of China (81270252 and 81700305).