
Abstract
A humoral mechanism of cardioprotection by remote ischemic preconditioning (RIP) has been clearly demonstrated in various models of ischemia–reperfusion including upper and lower extremities, liver, and the mesenteric and renal arteries. A wide range of humoral factors for RIP have been proposed including hydrophobic peptides, opioid peptides, adenosine, prostanoids, endovanilloids, endocannabinoids, calcitonin gene-related peptide, leukotrienes, noradrenaline, adrenomedullin, erythropoietin, apolipoprotein, A-I glucagon-like peptide-1, interleukin 10, stromal cell-derived factor 1, and microRNAs. Virtually, all of the components of ischemic preconditioning’s signaling pathway such as nitric oxide synthase, protein kinase C, redox signaling, PI3-kinase/Akt, glycogen synthase kinase β, ERK1/2, mitoKATP channels, Connexin 43, and STAT were all found to play a role. The signaling pattern also depends on which remote vascular bed was subjected to ischemia and on the time between applying the rip and myocardial ischemia occurs. Because there is convincing evidence for many seemingly diverse humoral components in RIP, the most likely explanation is that the overall mechanism is complex like that seen in ischemic preconditioning where multiple components are both in series and in parallel and interact with each other. Inhibition of any single component in the right circumstance may block the resulting protective effect, and selectively activating that component may trigger the protection. Identifying the humoral factors responsible for RIP might be useful in developing drugs that confer RIP’s protection in a more comfortable and reliable manner.
Introduction
Several forms of endogenous protection against injury from ischemia–reperfusion (I/R) of the heart have been described. Ischemic preconditioning (IPre) occurs after a brief period of I/R prior to the prolonged index ischemia. The cardioprotective effect of IPre against infarction is due to protective signal transduction pathways within the heart itself. In ischemic postconditioning (IPost), the protective signaling of IPre can be instated by starting the reperfusion phase with a series of very brief I/R cycles. In remote IPre (RIP) or remote IPost (RIPost), protection against injury from I/R is instituted by performing cycles of brief I/R on a vascular bed remote from the heart either before or after myocardial ischemia which then confers protection against infarction to the heart by an, as yet, incompletely understood mechanism. 1 -5 Early-phase RIP occurs when the protective effect manifests itself minutes after an RIP procedure, while in delayed or late-phase RIP a more long-lasting protection reccurs many hours after the RIP procedure has been performed. 6 -8 Two distinct pathways have been proposed for transmitting the protective effect of RIP: humoral and neurogenic. 9 -11 The preferential activation of one or the other according to Liem et al 12 depends on both the type of IPre, that is, which organ/tissue will undergo transient I/R and what the protective target might be (eg, arrhythmias or infarction). For example, the protective effect against infarction of the heart by kidney or intestinal I/R appears to be achieved primarily through the activation of afferent neural pathways, 13,14 whereas the cardioprotective effect after transient ischemia of skeletal muscles seems to involve both humoral and neurogenic pathways. 15 Because it is often difficult to clearly distinguish between humoral or nervous pathways in RIP, it is often referred to as simply “neurohumoral” factors. Nevertheless, the identification of humoral components of RIP as well as understanding their interaction with the heart could lead to treatments that increase the heart’s tolerance to I/R injury in clinical practice. 2
Humoral Factors
The first evidence that points to a humoral mechanism of the protective action of RIP was published by Oxman et al. 16 Researchers applied 10-minute ischemia followed by 10-minute reperfusion of the hind paw of a rat with an elastic cuff. The hearts were then removed and perfused with Krebs buffer. The isolated hearts experienced 30 minutes of regional ischemia followed by 15 of reperfusion. The RIP reduced the incidence of ventricular tachycardia during reperfusion by approximately 85%. 16
Proof that a humoral component is present in RIP comes for experiments like the following. Classical IPre of the heart (IPre) was performed with 5 brief episodes of occlusion/reperfusion of a coronary branch as well as of a renal artery. Subsequent cross-perfusion of whole blood between the donor rabbits with IPre and the recipient rabbits without IPre reduced the infarct size–area at risk (IS/AAR) ratio by approximately 75% after 1 hour of coronary occlusion and 30 minutes of reperfusion in both the donor and the recipient groups. When blood was similarly exchanged between non-IPre animals, there was no protection. 17 Later on, the same team would conduct a study of remote cardioprotective action of IPre in vitro. 18 IPre was performed with three 5-minute cycles of ischemia and reperfusion of the isolated rabbit heart, and coronary effluent was collected. This effluent was passed through acceptor hearts (without IPre), which significantly increased their mechanical function following a transient ischemic insult. 18 These studies confirm the presence of humoral factors that are produced in the heart of one animal experiencing IPre and exert their protective effect on the myocardium of another animal from a distance. Unfortunately, the authors did not identify these humoral factors, but they suggested that such mediators could be adenosine and/or norepinephrine. 18
Proteins
Bartekova et al studied the involvement of low-molecular-weight proteins in RIP using rat liver ischemia as the trigger. A low-molecular-weight fraction of proteins was isolated from the perfusion solution flowing from the isolated rat liver during its reperfusion. Perfusion of isolated rat hearts with a solution containing the isolated fraction of proteins increased their resistance to arrhythmias, improved postischemic recovery of contractile function, increased postischemic coronary flow, and increased content of ATP. 19 In their next article, they made the rat’s liver ischemic in situ by occluding the portal vein and the hepatic artery for 20 minutes and reperfused them for 30 minutes. The heart was then removed and perfused with Krebs buffer. Hearts from animals that had liver RIP had a better recovery of myocardial contractile function after global I/R than those without RIP. Electrophoresis of proteins in a polyacrylamide gel revealed a significant increase in the amount of low-molecular-weight fraction proteins in the plasma after the transient liver ischemia. 20 Clearly, the RIP had triggered a change in the heart’s tolerance to ischemia that persisted even when humoral factor in the coronary perfusate was no longer present. Bartekova et al did not determine the nature of these proteins, but, based on their in vivo and in vitro studies, low-molecular-weight proteins have been identified as a humoral component of RIP.
The coronary effluent from the isolated heart of a donor rat experiencing IPre had infarct-limiting and inotropic effects on the isolated heart of a naive recipient rat subjected to global I/R. 21 The coronary effluent was analyzed for the presence of humoral factors mediating the cardioprotection of IPre. First, the protective effect of the effluent was lost when protein kinase C (PKC) was blocked in the heart with chelerythrine. This indicates that the humoral mechanism of cardioprotection involves activation of PKC as occurs in IPre. 22 Secondly, they found that only the thermolabile hydrophobic fraction of proteins, with a molecular weight of more than 3.5 kDa, possesses the cardioprotective effect. 21
Konstantinov et al demonstrated that the cardioprotective effect of RIP is associated with the activation of ATP-dependent K-channels (KATP channels) and does not depend on the activation of the afferent nerves innervating the heart. Their experiment consisted of heart transplantation from donor pigs to recipient pigs who had previously experienced RIP by a tourniquet around the hind limb (4 cycles of 5-minute ischemia/5-minute reperfusion). Infarct size after coronary artery occlusion/reperfusion in the transplanted heart was reduced by 57%, 23 indicating that the humoral factor persists in the blood after RIP and does not require cardiac innervation. The infarct-limiting effect of RIP recipient animals was abolished after the blockade of the KATP channels by glibenclamide as is the case again with IPre. 22 The abovementioned data suggest an important role of proteins in RIP, but the molecular nature of these proteins is unknown.
Heat Shock Proteins
Another group involved in the search for humoral factors performed RIP with four 5-minute cycles of I/R of the back leg of the rabbit. Low-molecular-weight dialysate of plasma from these rabbits protected an isolated heart and that was shown to be due to hydrophobic low-molecular compounds in the plasma (<15 kDa). Nonselective blockade of opioid receptors with naloxone abolished its protection. 24 In 2017, Maciel et al 25 attempted to determine the molecular weight of the humoral factor by ultrafiltration. They used a coronary effluent from an IPre heart and used it to perfuse other hearts that underwent I/R. They also found that hydrophobic peptides with a molecular mass of 4 to 12 kDa exerted the infarct-limiting effect. One of these peptides was identified, and it turned out to be a heat shock protein (HSP-10). 25 Their cardioprotective effect was again associated with the activation of the canonical IPre signaling components PKC and KATP channel opening. 22
Opioids
The first evidence of the involvement of opioid peptides in the implementation of the cardioprotective effect of RIP was given by Dickson et al. 26 They perfused isolated rabbit hearts with a solution collected after IPre of isolated donor hearts. This reduced the IS–AAR ratio in response to 40-minute global ischemia and 120-minute reperfusion of the recipient hearts. 26 The addition of a nonselective opioid receptor antagonist, naloxone, to the perfusion solution eliminated the infarct-limiting effect of IPre. The humoral factors produced by the myocardium after IPre simulation had an antinecrotic impact not only on the myocardium itself but also on the other organs including the small intestine. The protective effect of this form of preconditioning was abolished by naloxone or blockade of KATP channels with glibenclamide. 27 The cardioprotective effect of RIP by occlusion/reperfusion of the mesenteric artery was also blocked with naloxone. 28 The antiapoptotic effect on the myocardium of RIP by occlusion/reperfusion of the femoral artery in pigs is also associated with activation of opioid receptors. The ganglion blocker hexamethonium before RIP did not reduce the antiapoptotic effect, which seemingly excludes any neurogenic mechanism of this model’s cardioprotective action. 29
Further research in this area has been aimed at identifying subtypes of opioid receptors, as well as their endogenous agonists, which participate in the mechanism of cardioprotective action of RIP. Weinbrenner and colleagues exposed an open-chest rat heart to 30-minute coronary occlusion and 30-minute reperfusion. The RIP was accomplished by I/R of the renal artery. The protective effect of RIP was eliminated by blockade of δ1 opioid receptors (OR) with BNTX. 30 Another group 31 identified κ OR in the cardioprotective mechanism. The RIP was induced in rats by 3 cycles of 5 minutes of right femoral artery occlusion followed by 5 minutes of reperfusion. The potency of RIP’s infarct size-reducing effect was comparable to that from selective agonists of the κ OR (U-50 488H, 10 mg/kg intravenously 30 minutes prior to coronary occlusion) or the endogenous κ OR agonist (dynorphin, 20 ng/kg intravenously 30 minutes before coronary occlusion). 31 Such a high biological activity of dynorphin (20 ng/kg) is a surprise because opioid peptides usually limit infarct size in a dose of several tens and hundreds of micrograms per kg. U-50 488H has a cardioprotective effect at a dose of 1 mg/kg. 32 The selective κ OR antagonist nor-binaltorphimine (10 mg/kg) completely abolished the protective effect of RIP and U-50 488H. Zhang et al 31 found a 3-fold increase in the concentration of dynorphin in the plasma 2 hours after RIP but no change in the endogenous δ OR agonist met-enkephalin. The selective blockade of δ OR did not affect cardioprotection after RIP. 31 The difference between the findings of Zhang et al and those of Weinbrenner et al is probably due to their use of very different RIP models.
Based on their previous studies on ORs in the implementation of IPre, Dickson et al 26,27 considered the peptide Met 5 -enkephalin-Arg 6 -Phe 7 (MEAP) as a possible humoral mediator. They found that intramuscular injection of MEAP (25 mg) 24 hours before 25-minute global ischemia and 120-minute reperfusion of an isolated rabbit heart reduced the IS–AAR ratio by approximately 60%. 33 The ability of MEAP to provide a second window of cardioprotection allows one to consider the participation of this peptide as a potential humoral factor in RIP. It was subsequently demonstrated that treatment of isolated rabbit cardiomyocytes with blood plasma dialysate, obtained after performing RIP with four 5-minute cycles of ischemia /5-minute reperfusion of a lower extremity of a donor rabbit, the percentage of necrotic death of cardiomyocytes after anoxia/reoxygenation was reduced by approximately 30%, and the selective blockade of δ OR with naltrindole (10 nmol/L) or the κ OR with GNTI (1 nmol/L) or adenosine receptors with 8-(p-sulfophenyl) theophylline (8-SPT) prior to anoxia/reoxygenation eliminated the cytoprotective effect. They failed to find elevated OR agonists, adenosine, or inosine in the plasma dialysate. It should be noted that protection from exogenous met-enkephalin (100 μmol/L) or dynorphin (100 μmol/L) could also be blocked with 8-SPT. 34 These data indicate the functional interaction of opioid and adenosine receptors was occurring in these myocytes. 35 Surendra and colleagues concluded that RIP cardioprotection requires the activation of δ-opioid and κ-opioid receptors and relies on these receptors functionally interacting with adenosine receptors. The substance in the dialysate that caused adenosine and opioid receptors to be activated in the Surendra et al study was not identified.
Using the example of the abovementioned studies, it can be concluded that endogenous opioid peptides and/or adenosine probably participate as local humoral autocoids in the implementation of the cardioprotective effect of RIP rather than a circulating hormonal role. The final decision on the participation of specific endogenous ligands of OR in the humoral mechanism of RIP is difficult to determine, since the participation of an opioid in RIP depends on the type of RIP used as well as on the chosen object of study.
Adenosine
There is considerable evidence that adenosine receptors are involved in the cardioprotective effect of RIP. 14,36 -39 The first evidence came from Pell et al in rabbits. The RIP was effected by I/R (10/10 minutes) of the left renal artery. The RIP caused a decrease by 50% in the infarct size in response to 30-minute coronary occlusion, 2-hour reperfusion of an in situ rabbit heart. 8-SPT, anon-selective adenosine receptor antagonist, eliminated the ability of the RIP to limit the size of the infarction. 36 Thus, the authors concluded that adenosine receptors are involved in the implementation of the infarct-reducing effect of renal RIP.
In a subsequent in situ rabbit study, either renal RIP or classical IPre reduced infarct size resulting from a 40-minute coronary occlusion followed by 2-hour reperfusion. The phosphocreatine and ATP content in the myocardium as measured by nuclear magnetic resonance spectroscopy was also preserved in the protected hearts. The 8-SPT prior to coronary occlusion eliminated protection from both RIP and IPre. 37 This reveals that as in classical IPre, renal RIP limits infarct size in the heart with the participation of adenosine receptors. It should be noted that adenosine’s involvement in IPre is complex. The IPre has both a trigger phase where a variety of G protein-coupled receptors can trigger the onset of protection. Regardless of the receptor that triggers the conditioned state, that protection is then mediated early in reperfusion by a complicated signaling pathway within the heart muscle that involves endogenous nitric oxide synthase, KATP channels, redox signaling, PKC, Akt, ERK, and adenosine receptors. 40 Thus, the adenosine may not be acting as a hormonal signal between the kidney and the heart, but it could be a local autocoid in conditioning’s mediator phase. Similarly, adenosine released from the ischemic kidney may not have traveled to the heart but rather could have activated a neural signal.
Liem et al explored the latter possibility. The RIP was induced by occlusion/reperfusion (15/10 minutes) of a rat’s mesenteric artery. This effect reduced the IS–AAR ratio by approximately 30%, and 8-SPT completely eliminated the infarct-limiting effect. Infusion of adenosine (10 μg/min) into the upper mesenteric artery had a cardioprotective effect comparable to that of RIP, which could be eliminated by the ganglion blocker hexamethonium. Infusing adenosine into the portal vein was not protective, proving that the infused adenosine was not acting directly on the heart but rather somewhere in the mesentery. Giving hexamethonium 5 minutes after reperfusion did not block protection from mesenteric artery adenosine but 8-SPT did. 14 Collectively, these data indicate that occlusion of the superior mesenteric artery causes activation of mesenteric adenosine receptors that stimulate afferent mesenteric nerve fibers that lead to a neural signal to the heart, the neural signal puts the heart into a protected state. The protective mechanism in the heart involves adenosine receptor activation early in reperfusion just as occurs in IPre’s mediator phase.
Dong et al reported a similar behavior with lower limb RIP in a rat model. Femoral artery occlusion or infusion of adenosine into the femoral artery reduced infarct size in the ischemia–reperfused heart. However, infusion into the femoral vein was not protective. 38 That again argued against adenosine acting as a hormonal mediator. Schulte et al investigated delayed RIP, which was carried out by transient ischemia of the brain of the mouse by bilateral ligation of the internal carotid arteries. 39 The concentration of endogenous adenosine in the blood after RIP increased by 2.5-fold 30 minutes after carotid artery ligation. Twenty-four hours later, the heart was isolated and subjected to I/R and was found to be protected. The cardioprotective effect of RIP was not present in adenosine A1 receptor deficient (A1R−/−) mice. It should be noted that the half-life of adenosine in whole blood is less than 10 seconds, 41 so it is unlikely that the adenosine released by RIP would still be present the next day. Yet adenosine was still required for protection, presumably as a mediator rather than a trigger. All of this casts doubt on adenosine’s role as a humoral factor in RIP. Adenosine does appear to take part in mediating the heat’s tolerance to I/R once it has received the protective signaling much as occurs in IPre. 40 The infarct-limiting effect of adenosine is associated with the activation of kinases (MEK1/2, ERK1/2, Akt, tyrosine kinase, PKC) and KATP channels. 5
Prostanoids
The first group to consider prostanoids in the implementation of RIP was Oxman et al. 16 The RIP was produced in rats by hind limb I/R followed by removal of the heart and perfusing it ex vivo. While the isolated hearts were protected against a regional I/R insult, the percentage of changes in the concentration of prostacyclin did not differ from those in the group of animals with RIP. They were unable to identify a relationship between the dynamics of prostacyclin release during reperfusion from rat myocardium after hind limb RIP. 16
Sharma et al 42 studied of the role of thromboxane A2 in the implementation of RIP. The RIP was effected with four 5-minute cycles of inflation/deflation of a cuff around a hind paw of the rat. The RIP limited the myocardial infarct size and improved its postischemic recovery of ventricular contractile function in response to 30 minutes of global ischemia followed by reperfusion. 42 Ozagrel (thromoboxane synthase inhibitor) or seratrodast (thromboxane A2 receptor antagonist) was administered intraperitoneally 30 minutes prior to RIP simulation. Inhibition of thromboxane synthase, as well as blockade of thromoboxane A2 receptors, eliminated protection from RIP, which suggests the involvement of thromboxane A2 in signaling in the cardioprotective effect of RIP. 42 It should be noted that the role of thromboxane A2 in RIP has not yet been confirmed by any other studies. Its half-life in the blood is 30 to 47 seconds, 43 so it could possibly serve as a humoral factor of RIP.
Cannabinoids and Endovanilloids
The participation of endogenous cannabinoids and cannabinoid (CB) receptors in the mechanism of RIP was studied by Hajrasouliha et al. 44 The RIP was accomplished by a transient 15-minute occlusion of a rat’s mesenteric artery. Blockade of CB2 receptors with AM630 (1 mg/kg) 15 minutes before RIP eliminated its infarct-limiting and antiarrhythmic effects, but the CB1 receptor antagonist AM251 had no effect on RIP’s protection. 44 Thus, endogenous agonists of CB2 receptors also appear to participate in the cardioprotective effect of RIP. Unfortunately, the authors did not measure endogenous cannabinoids in the serum in these rats.
Another group of humoral factors mediating the cardioprotective effect of RIP are endovanilloids, the agonists of transient receptor potential channels for vanilloid (TRPV) channels. The TRPV channels are mechanosensitive ion channels that conduct Na+ and Ca2+. 45 They are located on the sarcolemma of cardiomyocytes, 46,47 on endothelial cells of the aorta, atria, ventricles, and coronary arteries, 48 -50 and on the endings of the afferent nerves in the heart, skeletal muscles, and other organs. 51 -53
Randhawa and Jaggi 54 used four 5-minute cycles of inflation/deflation of a cuff located on a hind limb of a rat and then isolated its heart. The RIP improved the postischemic recovery of myocardial contractility and reduced the infarct size. The RIP’s protection could be blocked by treating the rats with the nonselective TRPV blocker ruthenium red to assess the participation of TRPV channels in the RIP’s protective mechanism. The study showed that the blockade of TRPV channels eliminated the antinecrotic, inotropic, and infarct-limiting effect of RIP. 54,55 The same effect was seen with the TRPV1 channel antagonist capsazepine. 56 The TRPV1 channel activator capsaicin also mimicked the cardioprotective effect of RIP, 57 which suggests that the activation of TRPV1 channels by endovanilloids plays an important role in the cardioprotective effect of remote IPre.
Some endogenous cannabinoids, for example, anandamide and 2-arachidonoylglycerol, are also endovanilloids. 58 Data on the pharmacokinetics of anandamide are not available, but it is known that its hypotensive effect persists for several minutes after intravenous administration. 58 It was found that 5 minutes after the addition of 2-arachidonoylglycerol to the blood plasma, its plasma level is about 10% of the initial amount. 59 The presented data indicate that in endocannabinoids with intravenous administration, sufficient time is available to exert a significant effect on the heart. This would be especially applicable if the remote organ secreted endocannabinoids for 5 to 10 minutes.
Thus, in spite of the paucity of supporting data that the formation of the cardioprotective effect of RIP depends on the activation of cannabinoid and vanilloid (TRPV1) receptors, one should not discount of the possible role these receptors could play as humoral mediators of RIP’s cardioprotection.
Erythropoietin
Erythropoietin (EPO) is a glycoprotein consisting of 165 amino acid residues and is synthesized mainly in the kidneys. 60 About 90% of the EPO circulating in the blood is of renal origin. 61 Therefore, there is every reason to label it as a hormone, but some authors consider it to be a cytokine as well. 62 The EPO administered just prior to reperfusion reduced infarct size in isolated perfused rat heart as well as in open chest in situ hearts. It was found that this effect was dependent on the activation of ERK1/2 and phosphoinositide 3-kinase (PI3K) kinases. 63 In a study by Oba et al, 64 RIP was induced in mice with 4 cycles of 5-minute ischemia/5-minute reperfusion of the right lower limb. One hour after RIP, the level of EPO in the serum of mice increased by about 40%, and EPO messenger RNA (mRNA) in the kidneys was increased several fold. The RIP reduced the infarct size by 33%. The infarct-limiting effect of RIP was abolished by anti-EPO antibodies. Renal denervation also attenuated the infarct-limiting effect of RIP and eliminated the rise in serum EPO. 64 These data strongly suggest that erythropoietin is one of the humoral factors of RIP. The renal nerve apparently mediates an increase in renal secretion of EPO in response to I/R of the limb. This view is shared by other researchers. 65
Apolipoprotein A-I
Rats subjected to 10-minute limb ischemia followed by 10-minute reperfusion had a 40% rise in apolipoprotein A-1 (ApoA-1) in their blood, and infarct size was reduced by 18%. Conversely, injection of ApoA-1 reduced infarct size by a similar amount. 66 ApoA1 heterozygous (ApoA1 (+/−)) and ApoA1 null (ApoA1 (− / −)) mice had a 25% and 50% increase in infarct size, respectively, over that in the wild-type mice indicating that endogenous ApoA-1 defends the heart against infarction. 67 In another study, injection of 10 mg/kg ApoA-I decreased the infarct size by 17%. 68 But because exogenous ApoA-1 has such a modest infarct-limiting effect, it seems unlikely that it plays a leading role in RIP. However, studies performed on ApoA1-deficient mice demonstrate that ApoA-1 still plays an important role in regulating the heart’s tolerance to I/R.
Glucagon-Like Peptide 1
The RIP with a 15-minute occlusion of both femoral arteries followed by reperfusion in rats led to a 50% reduction in infarct size, and blockade of the glucagon-like peptide-1 receptor (GLP-1R) eliminated the protection. 69 A GLP-1R agonist mimicked the infarct-limiting effect of RIP. However, RIP did not significantly increase GLP-1 in the plasma. Apparently, GLP-1 is not a humoral factor for RIP, but it might be useful for protecting the heart. Others share that view. 65
Interleukin 10
Mice experienced RIP with 3 cycles of 5-minute femoral artery occlusion / 5-minute reperfusion 24 hours prior to experimental myocardial infarction. Interleukin-10 was elevated in both the cardiac tissue and the blood in the RIP mice, while infarct size was reduced by 67%. These effects were blocked by anti-IL-10 receptor antibodies. The IL-10 knockout mice did not observe a similar decrease in the IS–AAR ratio. Injection of IL-10 reduced the infarct size by 80% and that protection could also be blocked by Anti-IL-10 receptor antibodies. 70 The same group subsequently found that knocking down hypoxia-inducible factor (HIF) 1α in heterozygous mice with a knockout allele blocked the next day rise in IL-10 and its cardioprotection. Administration of a recombinant adenovirus encoding an HIF-1α into mouse hind limb muscle cells increase IL-10 levels and decrease myocardial infarct size in the knockdown mice. 71 Their data indicate that IL-10 is involved in the second window of RIP and that its secretion is regulated by HIF-1α. However, its role in the early stage of RIP remains unclear.
The Chemokine Stromal Cell-Derived Factor-1α
Stromal cell-derived factor 1 (SDF-1α or CXCL12) is a chemokine of 10 kDa that is synthesized in tissues in response to hypoxia. 65 Its effects are associated with the activation of its receptor CXCR4. Hind limb RIP in rats caused a 50% increase in the level of SDF-1α in the blood plasma and cut the infarct size in half. AMD3100, a highly specific antagonist of CXCR4, eliminated the infarct-limiting effect of RIP. 72 Therefore, SDF-1α is yet another candidate for humoral factor in RIP.
Calcitonin Gene-Related Peptide
Calcitonin gene-related peptide (CGRP) is a member of the family of calcitonin peptides. It is found mainly in capsaicin-sensitive nerve endings and exists in 2 isoforms α-CGRP and β-CGRP. 73,74 The receptors for calcitonin gene-linked peptide, CGRP1 and CGRP2, belong to the family of G-protein-conjugated receptors and are found in a variety of organs, including the nervous tissue of the brain and spinal cord as well as on endothelial cells of coronary arteries and in both atrial and ventricular cardiomyocytes. 75
Release of CGRP from afferent nerve fibers is triggered by various factors, including ischemia, a decrease in pH, or lactate acidosis. 76,77 The CGRP release from afferent nerve terminals can also occur in response to stimulation of TRPV1 channels. 78,79 In particular, D’Alonzo et al 80 showed that the perfusion of the isolated rat heart with a solution containing 30 μmol/L capsaicin, which is capable of stimulating the release of CGRP from capsaicin-sensitive nerve endings, has an antiarrhythmic effect and promotes a better recovery of myocardial contractility after global I/R in vitro. Blockade of CGRP receptors by the CGRP antagonist CGRP8-37 eliminates cardioprotection from classical IPre. 81 The involvement of CGRP as a humoral factor in the cardioprotective effect of RIP was demonstrated by Wolfrum et al. 82 The RIP was induced in rats with 15-minute mesenteric artery occlusion and subsequent reperfusion for 15 minutes. The RIP decreased the infarct size ratio by approximately 75% after 30-minute coronary occlusion and 150-minute reperfusion in vivo, and the protection was lost after treatment with the selective CGRP antagonist CGRP8-37. The serum CGRP concentration after RIP was increased by 40% over that prior to RIP. It should be noted that the ganglion blocker hexamethonium did not affect the rise in serum CGRP.
Singh et al 55 also examined the role of CGRP in the cardioprotective effect of RIP. The RIP was performed with 4 cycles of 5-minute ischemia / 5-minute reperfusion of the hindlimb in rats, after which the heart of the animal was isolated and subjected to global ischemia (30 minutes) and reperfusion (120 minutes). 55 The RIP caused a decrease in the IS–AAR ratio and improved post-ischemic recovery of myocardial contractility and also had an antinecrotic effect after I/R of the isolated heart in rats. 55 Those cardioprotective effects of RIP were absent when sumatriptan was present to block the CGRP release. 55 The CGRP receptor antagonist CGRP8-37 (1 mg/kg) also eliminated the cardioprotective effect of RIP. 79 Together, the data support the involvement of CGRP as a humoral factor in the cardioprotective effect of RIP.
Leukotrienes
Receptors for leukotrienes are found in various organs, such as heart, bronchi, spinal nerves, and intestines. 83 -85 In one study, RIP was performed in rats by 4 cycles of 5-minute ischemia / 5-minute reperfusion of the animal’s hind limb with an inflatable cuff. 86 The heart was then removed and perfused ex vivo where it underwent 30 minutes of global ischemia / 120 minutes of reperfusion. The RIP caused a decrease by approximately 75% in the IS–AAR ratio and improved post-ischemic recovery of contractility. The RIP-induced cardioprotection was abolished by intraperitoneal administration of montelukast, a receptor antagonist of cysteine leukotrienes LTS4, LTD4, and LTE4 receptors prior to RIP. A similar blocking effect was provided by the 5-lipoxygenase inhibitor zileuton which was administered at 2.5 or 5 mg/kg intraperitoneally 30 minutes before RIP. 86 These data suggest that leukotrienes may also be involved in RIP’s cardioprotective effect. The half-life of leukotriene C4 in the blood is 79 seconds. 87 Consequently, there is reason to believe that leukotrienes could also act as humoral factors involved in the implementation of the protective action of RIP.
Norepinephrine
The RIP hearts had a 150% increase in norepinephrine concentration in the coronary effluent at the end of reperfusion. 18 The antiarrhythmic effect of RIP was lost after the administration of sympatholytic agent reserpine to rats (0.15 mg/kg, intraperitoneally 24 hours prior to study). 18 Thus, norepinephrine is one of the possible neurohumoral mediators through which the protective effect of RIP on the heart is realized. Norepinephrine as a humoral factor in RIP was first proposed by Dickson et al. 18 Indeed, norepinephrine titers in the blood increase after RIP. 88 It should be noted that the stimulation of β-adrenergic receptors in the period preceding ischemia can promote an increase in cardiac tolerance to I/R, 89,90 so norepinephrine cannot be discounted as both a neurogenic and a humoral factor in RIP.
Adrenomedullin
A study on the effects of rat limb RIP on I/R of the kidney examined adrenomedullin. Twenty four hours after RIP, the level of adrenomedullin in the blood of rats was increased along with protection against ischemic injury to the kidneys. 91 Adrenomedullin is a known cardioprotectant 92 and could possibly be involved in the protection. However, it should be noted that the kidneys were also protected immediately after RIP, but no change in the concentration of adrenomedullin in the blood of those rats was detected. 91 Unfortunately, the available evidence for adrenomedullin as the humoral factor is only based on correlation and that correlation is only seen with delayed-phase RIP.
Glycine and Kynurenine
Glycine is yet another candidate for the role of the humoral factor involved in RIP. In pigs, hind limb RIP resulted in a 10-fold increase in the level of glycine in blood plasma. Plasma dialysate from the pigs was then used to perfuse an isolated murine heart subjected to I/R. The dialysate from RIP pigs caused a decrease in the IS–AAR ratio by 18%, and the same effect was provided by glycine. Blocking glycine receptors with strychnin, eliminated the protective effect of the dialysate or exogenous glycine. 93 However, the weak infarct-limiting potency of glycine, in our opinion, argues against glycine alone as a humoral factor.
Metabolites in serum from rats was analyzed by a metabalomics approach, and glycine and kynurenine titers were strongly correlated with RIP. 94 In humans, RIP produced by 3 cycles of 5-minute ischemia /5-minute reperfusion of an arm caused modest (<10%) but significantly increases the plasma concentration of glycine, kynurenine, spermine, carnosine, and serotonin. Kynurenin alone reduced the infarct size in rats by approximately 30%, while glycine alone reduced it by only 15%, and combining the 3 was no more potent than kynurenin alone. 95 While kynurenine seems the more likely candidate for the humoral factor, this is based only on correlation.
Exosomes and MicroRNAs
MicroRNAs (miRNAs) are another group of humoral factors that have been proposed to mediate the cardioprotective effect of RIP. MicroRNAs are small noncoding RNA sequences (20-23 nucleotides) that suppress the expression of various genes in eukaryotic cells. 96 They occur as a result of RNA transcription by RNA polymerase II enzyme 97 with sequential processing of primary transcripts by Drosha and DGCR8 complexes inside the cell’s nucleus. 98 The MiRNA precursors (pre-miRNA, ∼70 nucleotides) migrate from the nucleus to the cytoplasm along with the exportin-5 protein. 97 In the cytoplasm, pre-miRNA is hydrolyzed with RNase III and then bound to the RISC (miRNA-induced silencing complex) complex, forming a mature miRNA molecule. According to some investigators, 99,100 the mature miRNAs together with a complex of RISC proteins bind to complementary sites on mRNA which blocks its translation. This allows miRNAs to participate in the regulation of a number of cellular functions and processes, such as growth, differentiation, proliferation, metabolism, and apoptosis. 101,102 It is generally believed that miRNAs achieve their protective effect by migrating with the blood flow to target cells inside exosomes. 103 Protective exosomes, in turn, can be released from cells whose tissue have undergone transient I/R. 103
Exosomes (also known as microvesicles) have a cup-like shape, are 50 to 150 nm in diameter, and can be secreted by the majority of mammalian cells. 104,105 Exosomes provide intercellular signaling and are contained in all body fluids (blood plasma, cerebrospinal fluid, saliva, urine, etc). 105,106 Exosomes carry lipids, proteins, 106 mRNA, 107 and miRNAs. 108 -110 In the heart, sources of exosomes include cardiomyocytes, 111 fibroblasts, 112 and vascular endothelial cells. 113
A perfusion solution flowing from the isolated hearts of donor rats subjected to IPre (3 cycles of 5-minute global ischemia/5-minute reperfusion) was able to protect a second isolated heart against global I/R. When exosomes were removed from the coronary effluent of the IPre hearts by centrifugation, the protection was lost. 114 In addition, immunoblot analysis using heat shock protein 60 as an exosome marker revealed that IPre increases the exosomes production of the heart. 114 In another study, serum was collected from rats receiving RIP by bilateral femoral artery I/R, and an exosome-enriched fraction of the serum increased the tolerance of HL-1 cardiomyocytes to hypoxia/reoxygenation, but enriched serum from non-RIP rats was not protective. 115 Intramyocardial injection of concentrated exosomes from hind limb RIP rats decreased in the IS–AAR ratio and improved pumping function of the heart while that from non-RIP rats did not. Additionally, an miRNA-24 antagonist blocked the protection from the RIP exosomes suggesting that miRNA-24 in the exosomes was responsible for the protection In addition, the RIPre exosomes prevented apoptosis of cardiomyocytes. 116 These studies strongly implicate participation of exosomes in the cardioprotective effect of RIP.
In one study performed by Duan et al, 117 hind limb RIP or classical IPre of the heart was performed in rats and then the hearts were removed and perfused ex vivo. Infarct size was reduced after 30 minutes of global ischemia and 60 minutes of isolated rat heart reperfusion in the RIP and IPre hearts. Interestingly, RIP caused a decrease in serum miRNA-1, while IPre increased it and miRNA-24. 117 Thus, ischemic conditioning of different organs produces different responses.
In another study, it was established 118 that RIP in rats with hind limb ischemia limited infarct size after 35 minutes of coronary occlusion in vivo and reperfusion for 2 or 6 hours in vivo. After 2 hours of myocardial reperfusion, a decrease in miRNA-1 expression was found in the myocardium, but after 6 hours of reperfusion it increased. The expression of brain-derived neurotrophic factor (BDNF) is regulated by miRNA-1, but there was no correlation between the expression of miRNA-1 and the myocardial content of BDNF which is thought to possess a cardioprotective effect. 118 However, caution should be exercised in studying protein expression in largely infarcted tissue, since low expression at 6 hours may have simply been due to necrosis.
Li et al reported that RIP using 4 cycles of I/R of the lower extremity in mice or inflating a cuff on the lower limb in humans was accompanied by a significant increase in miRNA-144 in the blood plasma. 119 Thirty minutes of global ischemia and 60 minutes of reperfusion of the isolated mouse heart led to a significant decrease in miRNA-144 expression in the myocardium. RIP as well as intravenous siRNA-144 (8 mg/kg) had an infarct-reducing effect in mice, 119 and intravenous administration of the antisense oligonucleotide miRNA-144 eliminated the cardioprotective effect of both miRNA-144 and RIP. Along with the increased plasma miRNA-144 after RIP, Li and colleagues found an almost a 4-fold increase in the exosomes content of miRNA-144 precursor as well as the argonaute-2 miRNA carrier protein. 119 Therefore, miRNA-144 expression would appear to play an important role in RIP’s cardioprotection.
The RIP also induces a rise in the level of miRNA-21 in blood and urine in children. 120 The authors proposed that miRNA-21 might be protective, since it is associated with a change in p38 kinase expression. The RIP causes an increase in the level of siRNA-24 in exosomes isolated from the plasma of preconditioned rats. 116 In an in vitro study performed on H9c2 cardiomyoblasts, miRNA-24 was shown to inhibit H2O2-induced apoptosis and necrosis of the H9c2 cells. The investigators then found that the protective effect of miRNA-24 in rats undergoing 45-minute coronary occlusion and 1-day reperfusion was associated with downregulation of the proapoptotic Bim protein. 117 The role of miRNA-24’s in RIP’s antinecrotic effect remains unclear, as RIP can increase as well as decrease its production. 121
When RIP was produced using I/R of the hind limb, it caused a decrease in the miRNA-29a/b/c level in skeletal muscles. This miRNA inhibits the expression of an inducible NO synthase (iNOS). 121 The authors proposed that a decrease in the miRNA-29a/b/c level in the blood plasma promotes iNOS expression in remote organs which provides the protective effect. Thus, exosomes that contain various proteins as well as mRNA and miRNA may well comprise an important humoral component of the cardioprotective effect of RIP. As far as the molecular targets of miRNA are concerned, there is still no complete clarity, and further research is needed.
Late-Phase RIP
Above, we discussed the early phase of RIP. But, like IPre, there is also a late phase that occurs many hours after the RIP event, and it also seems to involve a humoral factor. In one study, donor rats experienced RIP or no RIP, and 48 hours later their plasma was transfused to recipient rats. Regional cardiac I/R was performed to generate an infarct. Saline was used as a control. The blood plasma of both the RIP and the nonconditioned rats had an infarct-limiting effect, but the blood plasma of the RIP rats was more effective if the infarct size was assessed 24 hours after transfusion. Transfusion with plasma of the RIP rats caused an increase in the phosphorylation of Akt and ERK1/2 in the myocardium of the rats 24 hours after transfusion but did not affect the phosphorylation of transcription factor STAT. 122 It is generally accepted that there are 2 signaling pathways that increase the cardiac tolerance to I/R from IPre through phophorylation: (1) Reperfusion-Induced Salvage Kinases (RISK), which includes PI3, Akt, MEK1/2, and ERK1/2 kinases and (2) Survivor Activating Factor Enhancement (SAFE), which includes the JAK kinase and STAT transcription factors. 123 -125 The authors concluded that their humoral factor activates RISK. Later, the same team of authors showed that plasma transfusion of preconditioned rats cannot only limit the size of the infarction but also reduces the occurrence of ventricular fibrillation in coronary occlusion and reperfusion. 126
Clinical data also indicate the existence of a protective humoral factor (s). 127 -129 An increase in the level of interleukin 1α in the blood of cardiac surgical patients after RIP was reported. 127 The isolated rat heart was perfused with a solution containing 0.5% of blood plasma from people subjected to RIP. 128 It turned out that the blood plasma of the young volunteers reduced the IS–AAR ratio by 34%, but the blood plasma from elderly volunteers did not affect the size of the infarction. 128 Ney et al attempted to find a humoral factor in cardiosurgical patients experiencing RIP. The RIP patients had an increase in the level of macrophage migration inhibitory factor (MIF) and stromal cell-derived factor 1 (CXCL12) in their serum. However, RIP had no cardioprotective effect in that study. 129 Consequently, CXCL12 and MIF are not likely to be humoral RIP factors.
A humoral pathway for the implementation of cardioprotection with RIP was demonstrated in various preconditioning models. Further studies of the mechanism for the development of heart RIP suggest that it may involve hydrophobic peptides, 25 endogenous opioids (opioid peptides), 26,27,30,31,33,34 prostanoids, 42 endocannabinoids 44 and endovanilloids, 54 calcitonin, a gene-related peptide 55,82 as well as adenosine, 14,36 -39 leukotrienes, 86 norepinephrine, 88 adrenomedullin, 91 glycine, 93 and microRNAs. 116 -120
Cellular RIP Targets
Much is known about the mechanism of classical IPre. Like RIP, IPre was shown early on to involve a number of signal transduction components. Later investigations showed that these formed a complex network of serial and parallel interconnections, all apparently converging on the mitochondrial transition pore. Inhibition of any of the links in this chain would block IPre’s protection. A number of studies have examined IPre’s signaling components to see whether any are present in RIP’s mechanism. Because these are intracellular, they would be targets of the humoral agents released by RIP.
Heme Oxygenase 1
Heme oxygenase 1 (HO-1) catalyzes oxidation of the heme molecule and generates CO, a known signal molecule for the generation of cyclic guanosine monophosphate (cGMP), and the antioxidant bilirubin. 130 The cGMP is known to increase the heart’s tolerance to I/R. 131,132 It was established that renal ischemia for 30 minutes caused a 3-fold increase in HO-1 mRNA level in the heart and remained at this level for 24 hours. 130 The protein and mRNA levels of HO-1 in the heart was increased after delayed RIP was induced by RIP of hind limb. 133 It has been demonstrated that infarct-limiting effect of delayed RIP is blocked by pretreatment with zinc protoporphyrin IX, an HO-1 selective inhibitor, suggesting that HO-1 may be involved in RIP. 133
Nitric Oxide Synthase
Nitric oxide synthase (NOS) plays an important role in IPre and the heart’s tolerance to I/R. 132 The RIP was induced in rats by intestinal ischemia. The rats’ hearts were then subjected to I/R 24 hours later, and those receiving RIP were protected. Administration of aminoguanidine or S-methylisothiourea, both inducible NO-synthase (iNOS) inhibitors, 60 or 30 minutes before myocardial ischemia, respectively, abolished the infarct-reducing effect of RIP. 134 In another study, RIP was induced by 6 cycles of a 4-minute ischemia / 4-minute reperfusion of the small intestine 24 hours before coronary artery occlusion and reperfusion. 8 The RIP’s infarct-reducing effect was completely abolished by pretreatment with the NOS inhibitor L-NAME. Mice (wild-types or with targeted deletions of the iNOS gene) experienced RIP of a hind limb, and 24 hours later their hearts were isolated and experienced global ischemia and reperfusion. 135 The RIP reduced infarct size in wild-type mice but in not the knockout hearts. Cardiac mRNA for iNOS in the wild-type mice was increased 24 hours after RIP. 135 In another study, rats subjected to 4 cycles of 5-minute occlusion / 5-minute reperfusion of both hind limbs showed a reduced infarct size immediately following the RIP, and L-NAME eliminated RIP’s infarct-limiting effect. An involvement of NOS in both early and delayed cardioprotection has been confirmed by other investigators. 70,136 However, evidence against a role for NOS in an early RIP also exists. 137 The reason for this discrepancy remains unclear.
Protein Kinase C
It is well known that PKC plays an important role in IPre and IPost. 132 Rats were subjected RIP of the superior mesenteric artery followed by coronary artery I/R. The PKC inhibitors chelerythrine and staurosporine abolished RIP’s infarct-reducing effect. 138 Chelerythrine completely eliminated protection from RIP and induced a translocation of PKCε from cytosol to the particulate fraction indicating activation of this kinase. 139 Chelerythrine also eliminated cardioprotection from RIP by infrarenal occlusion of the aorta. 15 An involvement of PKC in RIP was reported by another study. 140 The presented data indicate that PKC plays an important role in RIP.
Reactive Oxygen Species
There is evidence that reactive oxygen species (ROS) are also involved in RIP. 30,136,141,142 For example, RIP was induced by infrarenal occlusion of the aorta, and infarction of the heart was induced by a 30-minute regional ischemia followed by a 30-minute reperfusion. Infarct size was decreased by 63% in the RIP hearts. Pretreatment with the free radical scavenger N-2-mercaptopropionyl glycine (2-MPG) completely eliminated RIP’s infarct-reducing effect. 30 In another study, RIP has been shown to increase the level of ROS in the blood of rats. The 2-MPG eliminated this increase and abolished the infarct-limiting effect of RIP. 141 The ability of 2-MPG to eliminate the infarct-reducing effect of RIP was corroborated by other researchers. 143 The RIP markedly attenuates I/R injury of isolated perfused rat heart. 142 Administration of the antioxidant N-acetyl cysteine during RIP attenuated its cardioprotective effects in the isolated heart. It has been shown that RIP increases mitochondrial production of H2O2 which may be the source of the protective redox signaling. 136 Thus, there are convincing data to indicate that ROS are involved in the infarct-reducing effect of RIP.
Phosphoinositide 3-Kinase /Akt
It is well known that (PI3K and Akt kinase are involved in postconditioning and preconditioning. 132 Mice underwent hind limb RIP which decreased the IS–AAR ratio by 51%. Western blot showed that RIP was associated with activation of the PI3K/Akt signaling pathway. 144 Domestic pigs underwent hind limb RIP following coronary artery branch I/R. RIP reduced infarct size, and wortmannin, a PI3K inhibitor, eliminated RIP’s protection. 145 Activation of cardiac Akt and PI3K in response to RIP has been reported by several other investigators. 70,136,146,147
Glycogen Synthase Kinase 3β
Glycogen synthase kinase 3β (GSK3β) plays an important role in IPre and the regulation of the heart’s tolerance to I/R. 40,132 Phosphorylation of GSK3β leads to inhibition of mitochondrial permeability transition pore (MPT pore) opening which prevents ischemia-induced apoptosis and mitochondrial damage. 132 Hind limb RIP induced in mice using 4 cycles of 5-minute ischemia / 5-minute reperfusion decreased the infarct size following regional I/R and induced phosphorylation and inactivation of GSK3β. 144 In another study, RIP induced by 4 cycles of a 5 minutes of liver ischemia / 5 minutes reperfusion evoked cardiac GSK3β phosphorylation. 148 Other investigators have also reported an association between GSK3β and RIP. 149
Extracellular Signal-Regulated Protein Kinase 1/2
Extracellular signal-regulated protein kinase 1 and 2 (ERK1/2) contribute to cell survival in adverse conditions and participates in IPre and IPost along with Akt and PI3K as part of the RISK pathway. 132 The RIP was induced by 3 cycles of 5-minute blood pressure cuff inflation /5-minute deflation in patients undergoing coronary artery bypass grafting. Western blotting showed increased ERK1/2 activation by phosphorylation in cardiac biopsies. 150 The RIP induced by liver ischemia evoked phosphorylation of cardiac ERK1/2 in rats, and the selective ERK1/2 inhibitor U0126 abolished an antiarrhythmic effect of RIP in these hearts. 148 These data indicate that ERK1/2 is involved in RIP induced the heart’s tolerance to I/R.
Janus kinase
Activation of Janus kinase (JAK) predisposes an increase in the heart’s tolerance to I/R. 40,132 Pigs were subjected to hind limb RIP before coronary artery I/R. The selective JAK inhibitor AG490 abolished RIP’s infarct-reducing effect. 147 This result demonstrates an involvement of JAK in the cardioprotective effect of RIP. Other investigators corroborate this finding. 151
Mammalian Target of Rapamycin
It is known that mammalian Target Of Rapamycin (mTOR) is involved in the regulation of the heart’s tolerance to I/R. 152 The RIP was induced by 4 cycles of a 5 minutes of ischemia followed by a 5 minutes of reperfusion of limb in mice. 153 It was demonstrated that RIP decreases the activation of mTOR. This result shows that mTOR may also be involved in RIP.
Hypothetical End Effectors of RIP
Signal Transducer and Activator of Transcription
It is known that signal transducer and activator of transcription (STAT) plays an important role in IPre and postconditioning. 40,132,154 STAT3 regulates transcription and the functional state of mitochondria, and STAT1 and STAT5 regulate transcription. 132 JAK, which is activated by RIP, activates STAT1, STAT3, and STAT5. 132 The first report of activation of STAT1 and STAT3 with RIP was published by Huffman et al. 151 In a subsequent clinical study, RIP induced by 3 cycles of 5-minute upper arm ischemia / 5-minute reperfusion in patients undergoing coronary bypass surgery increased STAT5 phosphorylation in cardiac biopsies. 155 Stat5-cKO mice did not exhibit RIP-induced cardioprotection. 156 An involvement of STAT3 in mice undergoing RIP was also reported (Figure 1). 140
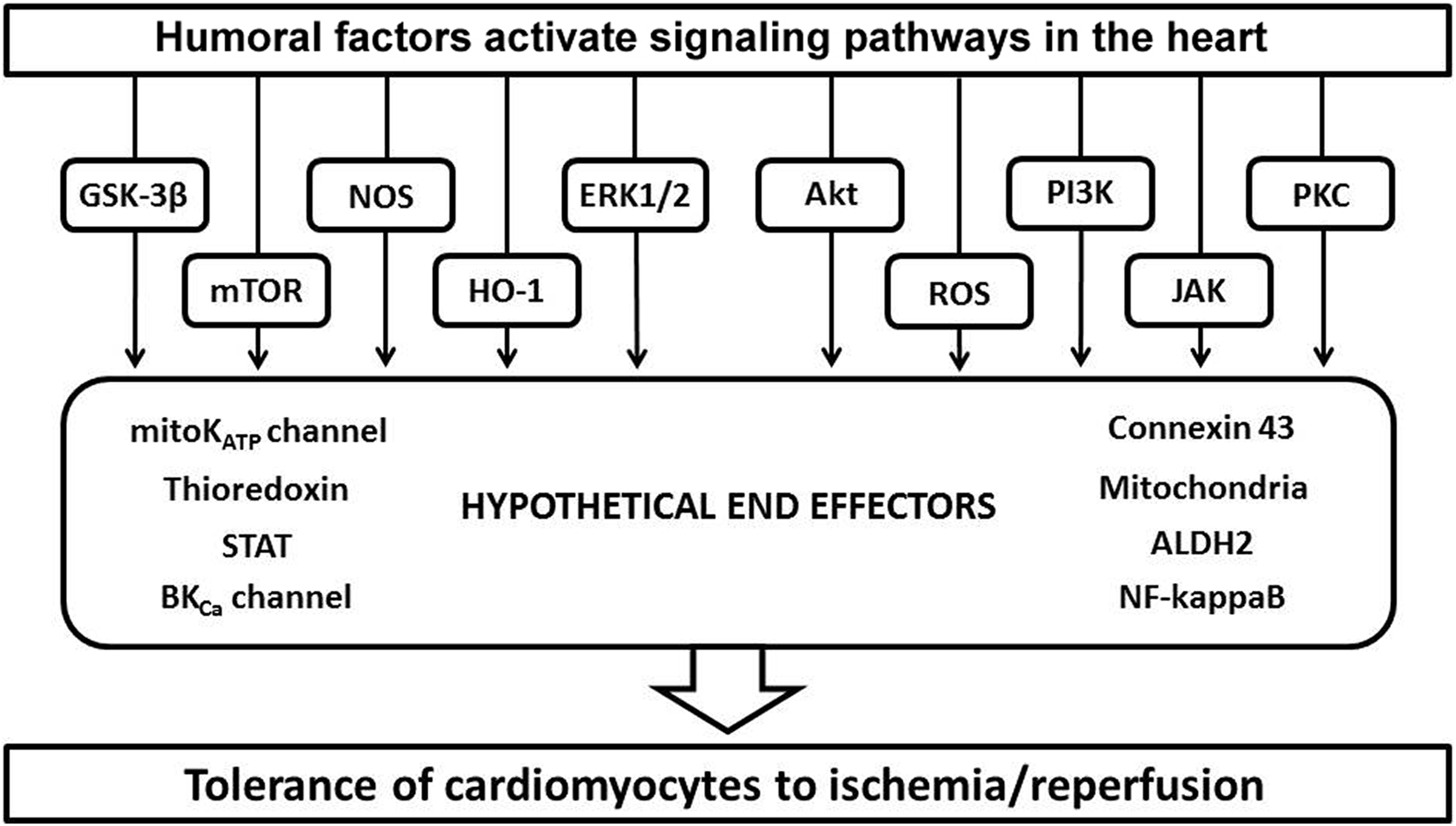
The cellular signaling pathways that the humoral factors were found to be dependent on to effect protection against cardiac ischemic injury. Note that these are identical to those seen in classical ischemic preconditioning.
Adenosine Triphosphate-Sensitive K+ Channels
It is well known that ATP-sensitive K+ channels (KATP channels) play an important role in IPre and postconditioning. 40,132 The RIP was induced with a 10-minute occlusion of the renal artery followed by a 10-minute reperfusion before coronary artery I/R in rabbits. RIP promoted a 46% decrease the infarct size. The selective mitochondrial KATP channel (mitoKATP channel) inhibitor 5-hydroxydecanoate eliminated RIP’s protection. 36 In another study, RIP was induced by hind limb I/R in rats. The hearts were then isolated and subjected to I/R. RIP reduced the heart injury and improved the left ventricular function during reperfusion compared to control. The nonselective KATP channel inhibitor glibenclamide and the selective mitoKATP channel inhibitor 5-hydroxydecanoate abolished RIP’s protection, but the selective sarcolemmal KATP channel inhibitor HMR-1098 had no effect. 157 An involvement of KATP channels in RIP has been confirmed by many others (Figure 1). 25,136,158 -160
Mitochondria
It is well known that mitochondria are hypothetical end effector of IPre and IPC. 132 It was appropriate to assume that they play an important role in the RIP. The first report on the possibility of mitochondrial participation in the RIP was published by Wang et al. 161 In a study that was performed on the cardiac surgery patients, RIP was performed preoperatively by using the inflating blood pressure cuff on the upper arm to 200 mm Hg for 3 × 5 minutes, with 5 minutes reperfusion intervals. 146,162 It was established that RIP promotes a preservation of maximal mitochondrial respiration. In our opinion, an additional study is needed to find out the role of mitochondria in the RIP (Figure 1).
Nuclear Factor Kappa B
It is known that nuclear factor kappa B (NFkappaB) is involved in the signaling pathway of late preconditioning. 162 -165 It could be assumed that this transcription factor is involved in late-phase RIP as well. Mice (wild-types, or with targeted deletions of the NFkappaB p105 gene) were subjected to 6 cycles of I/R of hind limb, and 24 hours later their hearts were isolated and subjected to global I/R. 135 adenosine triphosphate RIP reduced infarct size in the wild-type mice. Targeted deletions for the p105 subunit of NFkappaB eliminated an infarct-limiting effect of RIP. In our opinion, additional study is needed to further elucidate the role of NFkappaB in the RIP (Figure 1).
Big-Conductance Ca2+-Activated K+ Channels
It is well known that big-conductance Ca2+-activated K+ (BKCa) channels are localized on sarcolemma and in mitochondria. These channels are also involved in cardioprotective effect of IPre. 166,167 Consequently, it seemed likely that these channels may participate in the infarct-limiting effect of RIP. The RIP was induced by 4 cycles of 5-minute occlusion / 5-minute reperfusion of the left femoral artery. 168 The nonselective BKCa channel inhibitor paxilline eliminated the cardioprotective effect RIP, and the selective mitochondrial BKCa channel opener NS1619 could mimic RIP’s protection. More studies are needed (Figure 1).
Aldehyde Dehydrogenase
Aldehyde dehydrogenase (ALDH-2) eliminates aldehydes that are formed as a result of lipid peroxidation. There is evidence that this enzyme is involved in IPre. 169 The RIP was induced by 4 cycles of 5-minute hind limb ischemia / 5-minute reperfusion. The heart was then isolated and subjected to global I/R. The RIP decreased in the IS–AAR ratio by 43%. The ALDH inhibitor cyanamide eliminated RIP’s protection. 170 These data suggest that ALDH-2 also plays an important role in RIP. These data have not yet been confirmed by the other investigators (Figure 1).
Connexin 43
Connexin 43 is a member of the family of connexins, which are components of gap junctions, which form intercellular channels and allow the diffusion of low-molecular-weight compounds between adjacent cells. 171 Connexin 43 is involved in preconditioning and postconditioning. 132 RIP was induced by 4 cycles of 5-minute bilateral hind limb ischemia / 5-minute reperfusion in rats before coronary artery occlusion and reperfusion. RIP reduced the infarct size by 54%. I/R caused a strong decrease in connexin 43 expression in the AAR that was partly abolished by RIP. Furthermore, RIP decreased the ischemia-induced dephosphorylation of connexin 43 (Figure 1). 172
Thioredoxin
Thioredoxins are a group of proteins that protect cells from the toxic effect of ROS. 173 Thioredoxin-1 is involved in IPre. 174 A study was performed on cardiac surgical patients on cardiopulmonary bypass. The RIP was induced by four 5-minute cycles of upper limb I/R. The right atrial tissue was obtained from the patients who were receiving RIP and the control patients before and after cardiopulmonary bypass. It was shown that RIP promotes an increase in thioredoxin level in right atria by 25%. 175 This was only a correlation study. and further animal studies are needed to critically test whether the increase in thioredoxin is essential for RIP’s protection (Figure 1).
Blood plasma dialysate from rabbit donors who experienced RIP with 4 cycles of 5-minute ischemia / 5-minute reperfusion of the hind limb improved the post-ischemic function after cardioplegic arrest of a naive isolated rabbit heart. The RIP donor rabbit blood dialysate also increased the resistance of myocardial mitochondria to simulated ischemia. This was demonstrated by an increase in the rate of state 3 respiration (the rate of oxygen consumption after the addition of ADP to the incubation medium) and by maintaining the integrity of the outer mitochondrial membrane with a decrease in the release of cytochrome C. 161 Heusch group showed that the perfusion of the isolated rat heart with a solution containing the blood plasma of pigs after RIP promotes a decrease in the infarct size, increases the state 3 respiration of cardiac mitochondria, enhances their ATP synthesis, and reduces their production of reactive oxygen species. 1 In 2014, Leung et al 176 found that the protective effect of RIP on mitochondria of the myocardium was dependent on the activation of the heart’s adenosine receptors.
Conclusion
The cardioprotective effect of RIP clearly involves the release of circulating humoral factors that transfer the protective signal from the organ that underwent transient ischemia to the target organ (in particular, but not limited to, the heart). The strongest evidence of such a humoral component appeared in the experiments with cross-perfusion of the heart with blood from the donor animal undergoing RIP. 17 The humoral pathway of RIP has been demonstrated on various models of RIP. The strongest candidates for the humoral component of RIP include hydrophobic peptides, endogenous opioids, endocannabinoids, endovanilloids, prostanoids, norepinephrine, adrenomedullin, leukotrienes, as well as calcitonin gene-bound peptide and miRNAs as a component of exosomes (Figures 2 and 3).

The secondary humoral factors are involved in cardioprotective effect of remote ischemic preconditioning. The secondary humoral factors can play the role of intermediaries between the circulation humoral factors and a cell, like adenosine, or their role in the cardioprotective effect is questionable, such as glycine.

Humoral factors that have been implicated in remote ischemic preconditioning. The grouping shows possible interactions with each other.
There seems to be a strong overlap in the cellular signaling that mediates RIP’s protection and that for IPre suggesting that RIP effects its protection at the heart by the conditioning mechanism. Despite the obvious evidence of the involvement of adenosine receptor activation in the cardioprotection mechanism in RIP, adenosine may be involved in the mediator phase of protection 62 as is the case within the heart rather than as an external humoral trigger. The number of candidates for the humoral factor is surprisingly large. Logic would say that only one of them can be the real humoral factor but that may not be the case. Signaling in preconditioning turned out to be extremely complex with many components arranged both in series and in parallel with one another. That provided many points where an inhibitor would block preconditioning’s protection and an activator would mimic it. Much the same may be happening with RIP as the various humors and their targets interact with each other (Figure 1). Hopefully, future studies of the humoral factors involved in RIP will resolve the actual role of each of these agents and more importantly identify clinically useful treatments for protecting patient’s hearts.
Footnotes
Authors’ Note
The section dedicated to CGRP is framed within the framework of state assignments AAAA-A15-115120910024-0. The section on glycine and kynurenine was created with the support of Russian Foundation of Basic Research 18-415-700004.
Acknowledgment
The authors are grateful to D. Sipkova for technical assistance.
Author Contribution
Sergey Y. Tsibulnikov, Alexander S. Gorbunov, Nikita S. Voronkov, Alla A. Boshchenko, Sergey V. Popov, Ekaterina S. Prokudina, Nirmal Singh contributed to acquisition, analysis, or interpretation and contributed to conception or design. Leonid N. Maslov contributed to acquisition, analysis, or interpretation; drafted the manuscript; and agreed to be accountable for all aspects of work ensuring integrity and accuracy. James M. Downey contributed to acquisition, analysis, or interpretation; contributed to conception or design; drafted the manuscript; critically revised the manuscript; gave final approval; and agreed to be accountable for all aspects of work ensuring integrity and accuracy.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research and/or authorship of this article: The article was prepared with the support of the Russian Science Foundation grant 19-15-00037.