
Abstract
Background:
To evaluate the impact of atorvastatin discontinuation on the progression and stability of atherosclerotic plaques in a valid animal model of atherosclerosis.
Methods:
Seventy ApoE−/− male mice fed with high-fat diet were randomly assigned into: (1) long-term intervention groups: (i) ATL, received atorvastatin for 12 weeks, (ii) CO-12W, control received vehicle for 12 weeks, (iii) ATW-6W, received atorvastatin for 6 weeks which was withdrawn for another 6 weeks. (2) Short-term intervention groups: (i) ATS received atorvastatin for 6 weeks, (ii) CO-6W, control receiving vehicle for 6 weeks, (iii) ATW-3D, ATW-7D, received atorvastatin for 6 weeks which was withdrawn for 3 days and 7 days, respectively. Daily dosage of atorvastatin was 20 mg/kg. Mice were killed and aortic samples were obtained for histological evaluation.
Results:
Long-term atorvastatin treatment (ATL) induced atherosclerosis regression and stabilization compared to control (P < .05). Atorvastatin’s withdrawal was associated with acute (ATW-3D) reduction in connective tissue and collagen contents within plaques compared to ATS (P < .05). Those changes were almost restored after a while (ATW-7D) and started appearing again after longer cessation (ATW-6W). Moreover, atorvastatin withdrawal induced shortly (ATW-3D) a peak in inflammatory markers (macrophages, MCP-1, tumor necrosis factor-α) and matrix metalloproteinases (MMP-3, MMP-9) concentrations within plaques, which sustained but to a lesser extent along time (ATW-7D, ATW-6W).
Conclusion:
Short-term withdrawal of atorvastatin seems to compromise its antiatherosclerotic effects, leading to an unstable phenotype of the atherosclerotic lesions and a rebound increase in inflammatory mediators. The clinical relevance of our findings requires further investigation.
Introduction
Statins, a class of lipid-lowering drugs, 1 have emerged as an important pharmaceutical therapy for primary and secondary prevention of cardiovascular diseases. 2 –4 Those effects have been associated with the beneficial impact of statins on atherosclerotic plaque development and stability. 5,6 Regarding the underlying atheroprotective mechanisms, statins have shown, in addition to lipid-lowering, “pleiotropic” properties and anti-inflammatory and antioxidative effects. 7,8 In human and murine populations treated with statins, there is a strong association between the degree of inflammatory suppression (tumor necrosis factor-α—TNF-α, monocyte chemoattractant protein-1—MCP-1) and atherosclerotic plaque stabilization. 9,10 On the other hand, matrix metalloproteinases (MMPs) and their inhibitors (TIMPs-tissue inhibitor of metalloproteinases) have emerged as key factors in atherosclerosis progression. 11 Their expression is predominantly regulated by inflammatory factors. The authors and other investigators have demonstrated a statin-induced plaque stabilization via the favorable changes in MMP/TIMP concentrations within plaques. 12,13
Poor prognosis associated with low compliance to medical treatment is observed in many drug classes including statins. This is an important issue, not limited to financial reasons. 14,15 A growing body of evidence has described a rebound phenomenon after statin withdrawal, triggering acute cardiovascular events, such as myocardial infarction (MI), and leading to a significantly greater likelihood for hospitalization. 16,17 The Platelet Receptor Inhibition in Ischemic Syndrome Management (PRISM) study investigated the effects of statins on cardiac events rate in 1616 patients who had coronary artery disease and chest pain symptoms. 17 The interruption of statin after admission multiplied the cardiac risk during the 30-day follow-up compared to patients who maintained statin. Up to now, mechanistic explanations for potential adverse events, after statin withdrawal, have been proposed. Among them, the predominant hypothesis supports an exaggerated stimulation of pro-inflammatory pathways promptly after statin cessation in either stable (eg, hypercholesterolemic patients), 18 or unstable patients (eg, acute MI patients). 19 However, that evidence is based on changes in blood levels of inflammatory mediators and it remains unclear whether those changes affect the size and composition of atherosclerotic plaques, related to acute events incidence in patients previously treated with statins.
In the present study, we investigated the impact of atorvastatin discontinuation on the stability of atherosclerotic plaques along time, by assessing collagen and connective tissue contents. Furthermore, we searched for changes in inflammatory factors (MCP-1, TNF-α), MMPs (MMP-2, 3, 9) and their inhibitor (TIMP-2) within plaques and whether they were related to the structural changes in atherosclerotic plaques’ composition. For this purpose we used apolipoprotein double knockout (ApoE−/−) mouse a reliable and valid atherosclerotic animal model. 20,21
Materials and Methods
Study Design
All experiments were performed in the Biomedical Research Foundation of the Academy of Athens (BRFAA) and the protocol was evaluated and approved by the Veterinary Service of the Prefecture of Athens (permit number 2697/26-04-2013), as required by the Greek legal requirements for animal experimentation. The facility in BFRAA is registered as a “breeding” and “experimental” facility according to the Greek Presidential Decree 56/2013, which harmonizes national legislation with the European Community Directive 63/2010 on the Protection of Animals Used for Scientific Purposes.
Animals were housed in pairs in H-Temp polysulfone type III open top cages (425 mm (L) × 266 mm (W) × 185 mm (H); Tecniplast, Milan, Italy]. All cages were kept in the same animal room with High Efficiency Particulate (HEPA)-filtered air supply, 15 ACH (air changes per hour), at a room temperature of 24°C ± 2°C, relative humidity of 55% ± 10%, 12-h light/dark cycle (light on between 07:00 and 19:00 hours), light intensity of 300 lx (measured 1 meter above the floor in the middle of the room), and positive air pressure of 0.6 Pa within the room.
All mice had ad libitum access to filtered tap water in drinking bottles and to western-type, high-fat diet (HFD) that contained 42% of total calories from milk fat and 0.15% from cholesterol (Teklad TD 88137; Harlan, Italy).
A total of 70, 8 weeks old, ApoE−/− male mice were enrolled. They were fed HFD for the whole experimental period. On the 10th week of experiment, all mice were randomly assigned into the following 2 arms: Short-term intervention consisted of the following groups: (1) Short-term atorvastatin (ATS) group, received atorvastatin for 6 weeks; (2) control (CO-6W) group received orally vehicle daily for 6 weeks; (3) 2 atorvastatin withdrawal (ATW) groups: (i) ATW-3D group received atorvastatin for 6 weeks and then atorvastatin was withdrawn for 3 days; (ii) ATW-7D group received atorvastatin for 6 weeks and then atorvastatin was withdrawn for 7 days. Long-term intervention, which included the following groups: (1) Long-term atorvastatin (ATL) group, received atorvastatin for 12 weeks; (2) CO-12W group received orally vehicle daily for 12 weeks; (3) ATW-6W group received atorvastatin for 6 weeks and then atorvastatin was withdrawn for another 6 weeks. All groups were equal (n = 10; Supplemental material).
Daily dosage of atorvastatin (20 mg/kg), the dilution (water for injection) and the way of administration (esophageal gavage) were recommended by the manufacturer (Pfizer, New York City, New York). Mice body weight measurements were obtained before therapeutic interventions and at end point.
Intraperitoneal Glucose Tolerance Test
At the end of the experimental period, mice underwent an intraperitoneal glucose tolerance test after overnight fasting (6 hours). Conscious animals were administered, via intraperitoneal injection, glucose at a dose of 2 g/kg body weight (20% dextrose in phosphate-buffered saline [PBS]). Blood sampling from the tail was carried out at baseline, before glucose administration, and after 30, 60, 90, and 120 minutes while glucose levels were measured by Accu-Chek advantage glucose monitor (Roche Diagnostics, Indianapolis, Indiana).
Biochemical Measurements
Lipid profile (cholesterol and triglycerides levels) was assayed by an enzymatic method (Chemwell 2910; Awareness Technology Inc, Palm City, Florida). After overnight fasting, blood samples were obtained at the time of euthanasia, under isoflurane anesthesia (IsoFlo; Abbott, Illinois, USA) and via cardiac puncture. Blood samples were initially centrifuged at 3000 rpm for 15 minutes and then they were stored in deep freezer (−80 C) until analysis.
Tissue Collection and Processing
All mice were euthanized by exsanguinations after induction of anesthesia with isoflurane 2%. The heart and the aorta were washed thoroughly with PBS. Thereafter, the aortic root was excised and fixed in 10% buffered formalin overnight and then embedded in paraffin blocks.
Histomorphometric Analysis, Collagen, and Connective Tissue
Serial sectioning (5 μm thickness) at the level of aortic valve was performed with a microtome (Leica RM2255—Fully Automated Rotary Microtome, Leica Biosystems GmbH Wetzlar, Germany), starting when the leaflets became obvious. For each staining per mouse, 3 nonconsecutive aortic slices (at equal intervals of 50 μm) were mounted on poly-
Hematoxylin and eosin (H&E) stained slices were used for quantitative analysis of atherosclerotic burden. The extent of all atherosclerotic plaques (in mm2) was measured in each section and the total lumen area (in mm2) was calculated by measuring the internal elastic lamina perimeter and extrapolating the circumscribed area for each section. 12,22 We averaged plaque area and lumen area from all H&E stained sections per mouse, and the ratio of plaque area divided by the total lumen area was calculated and expressed as percentage of stenosis. The contents of collagen and connective tissue within the atherosclerotic plaques were quantified by histochemical staining of sections with Sirius Red and Masson’s trichrome, respectively. Image analysis software (Image Pro Plus version 4.1; Media Cybernetics; Rockville, Maryland, USA) was used for morphometric image analysis.
Immunohistochemical Analysis
Paraffin sections of the aortic valve were stained immunohistochemically with antibodies directed against Mac-3 antigen of murine macrophages (BD Pharmingen, Franklin Lakes, New Jersey), and smooth muscle isoform of actin (Biocare Medical, LLC, Concord, California). Nonconsecutive sections were also stained with polyclonal antibodies against MMP-2, MMP-3, MMP-9, TNF-a, MCP-1 (MBL International Corporation, Woburn, Massachusetts), and TIMP-2 (Acris Antibodies GmbH, Herford, Germany).
Each parameter was quantitatively assessed on 3 nonconsecutive sections of the aortic valve per mouse (10 mice per group). Results are given as percentage of the positively stained tissue area of the whole atherosclerotic plaque area.
Statistical Analysis
All data are expressed as the mean (standard deviation [SD]) and analyzed by SPSS (version 22.0; IBM Corp, Armonk, New York). Data analyses were performed using a paired t test to compare the differences within groups or a one-way analysis of variance with post hoc tests to compare the differences between groups at the end of the study. For comparison reasons, we used CO-6W as control group of the ATS, ATW-3D, ATW-7D groups, while CO-12W ran as control group of ATL and ATW-6W groups. A P value <.05 was considered statistically significant. For the glycemic profile, the trapezoidal rule was used to determine the area under the curve (AUC) after IPGTT.
Results
Body Weight, Biochemical Analysis, and IPGTT
All mice, independently from the group, showed increased weight at the end point. Pairwise comparisons revealed a statistically significant increase of body weight measurements from baseline to each end point (P < .05; data not presented).
Total cholesterol levels differed between groups at the end of the study. As expected, atorvastatin-treated mice exhibited decreased cholesterol levels when compared to the respective control groups (P < .05). Moreover, cholesterol values were significantly elevated in ATW-7D and ATW-6W groups compared to ATS and ATL groups, respectively (P < .05). No significant differences in triglycerides levels were detected between groups in each arm (P > .05).
As ‡ far as glycemic profile with GTT was concerned, control groups showed a larger AUC (CO-16W: 42335.25 ± 7313.33 mg min/dL and CO-22W: 44260.83 ± 8474.40 mg min/dL) compared to almost all treatment groups in each arm. Only ATW-6W ApoE−/− mice appeared with deteriorated glucose tolerance after 6 weeks of atorvastatin withdrawal than controls. Biochemical results are presented in Figure 1.

(A) Total cholesterol, (B) triglycerides, (C) glucose serum levels and (D) serum glucose AUC values, after glucose tolerance test. All values are expressed as mean (SD). *Statistically significant difference compared to CO-6W. **Statistically significant difference compared to CO-12W.
Statistically significant difference compared to both ATS and CO-6W.
 Statistically significant difference compared to both ATL and CO-12W.
Statistically significant difference compared to both ATL and CO-12W.
Morphometry, Collagen, and Connective Tissue
The effect of atorvastatin treatment on atherosclerotic lesion progression was evaluated by measuring luminal stenosis at the level of aortic valve. The changes in plaque stability were assessed by measuring collagen and connective tissue concentrations within the atherosclerotic plaques (Figure 2).

Representative pictures of the atherosclerotic plaque histomorphometric analysis: (A) hematoxylin/eosin staining for morphometry (upper panel); (B) Sirius red for collagen (middle panel); and (C) Masson’s Trichrome for connective tissue (lower panel) content (×20; scale bar: 0.25 mm).
The degree of luminal stenosis was significantly less in long-term atorvastatin-treated group (ATL) group (13.90 ± 1.08%) in comparison with its control group (21.95% ± 2.40% in CO-12W, P < .001). Notably, long-term (6 weeks) atorvastatin discontinuation almost reversed that beneficial effect of atorvastatin (ATW-6W vs ATL, P = .051). Short-term atorvastatin therapy (ATS) and the related 2 groups with discontinued atorvastatin treatment (ATW-3D, ATW-7D) appeared with less atherosclerotic burden than control group, but those differences did not achieve statistically significant levels (P > .05).
Sirius red staining revealed a statistically significant higher collagen content ATL (P < .001) and ATS mice (P < .05) than their respective controls. Besides this, long-term (6 weeks) atorvastatin withdrawal offset its impact on collagen concentration, since there was no statistically significant difference between ATW-6W and CO-12W (P > .05).
On the other hand, atorvastatin withdrawal acutely decreased collagen content (ATW-3D vs CO-6W, P = .001). That effect was transient and reversed 7 days later, since collagen concentrations elevated again in ATW-7D group compared to CO-6W (P < .05).
A similar pattern of changes in connective tissue contents within the atherosclerotic plaques was also noted. Both short and long-term atorvastatin therapy led to higher connective tissue content compared to the respective untreated animals. However, cessation of atorvastatin administration acutely, within 3 days, lowered connective tissue concentrations to control levels in relation to all other atorvastatin groups in the short-term arm (P < .001). All morphometric data are presented in Figure 3.
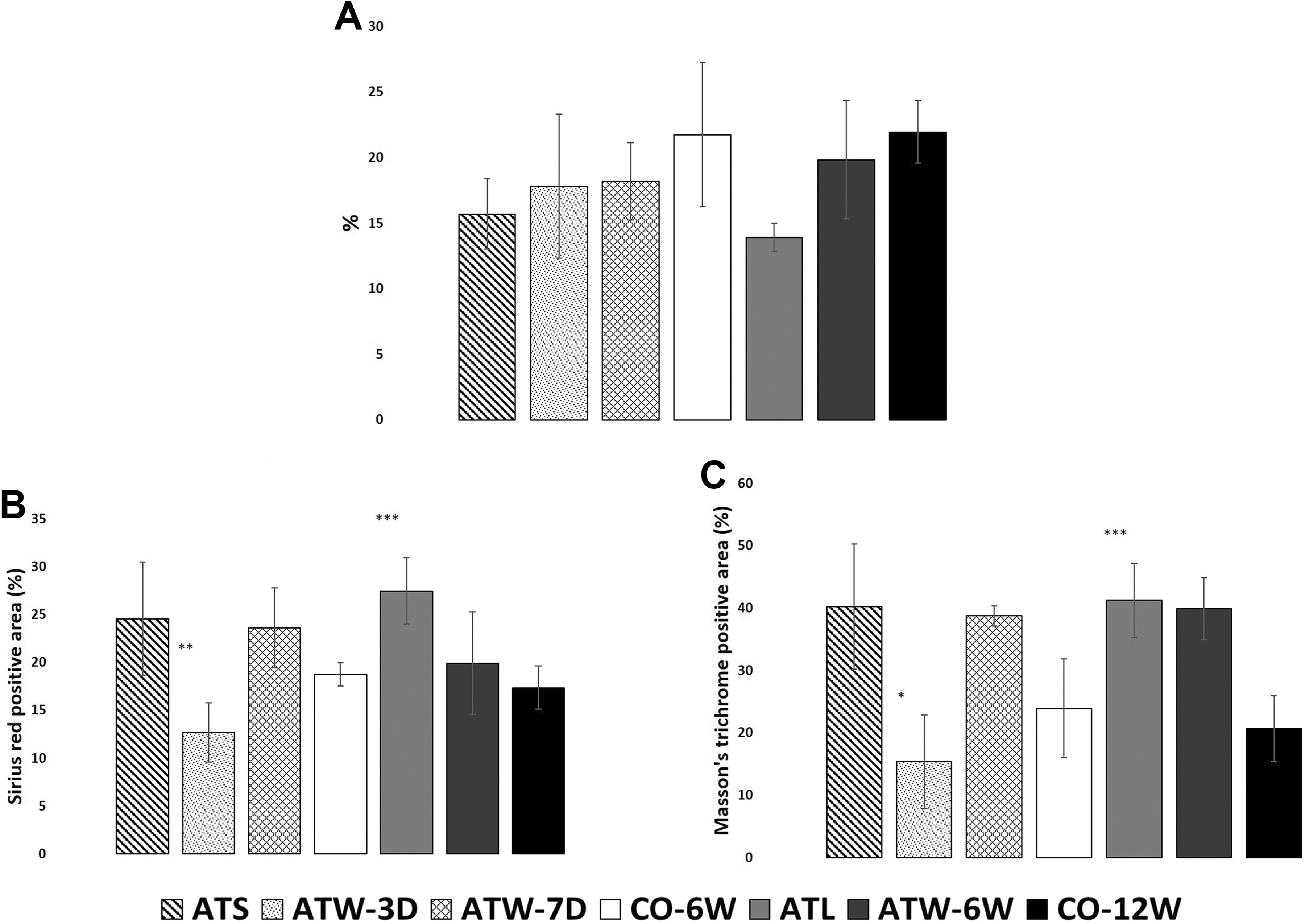
Histomorphometric results regarding (A) atherosclerotic stenosis percentage of the aortic root (H&E staining), (B) plaque collagen content (sirius red staining) and (C) connective tissue content (Masson Trichrome staining). All values are expressed as mean% (SD). *Statistically significant difference compared to ATS. **Statistically significant difference compared to both ATS and CO-6W. ***Statistically significant difference compared to CO-12W.
Immunohistochemistry
In comparison to respective control groups all atorvastatin-treated groups showed higher a-actin concentrations without achieving statistical significance (P > .05). The ATL and ATS groups exhibited a great reduction in the percentage of positive-stained area for macrophages compared to CO-12W and CO-6W, respectively (P < .05). Withdrawing atorvastatin treatment led after 3 days to a considerable increment in macrophage concentrations compared to ATS (ATW-3D: 20.85% ± 2.50%, ATS: 10.96 ± 4.46%, P < .05). Corresponding increase was also observed in ATW-6W in relation to ATL group but without any statistically significant difference (P > .05).
In the long-term arm of our study, 6 weeks withdrawal of atorvastatin (ATW-6W) was associated with a considerable increase in TNF-α, MCP-1, and all MMPmember concentrations, while TIMP-2 was significantly decreased compared to the relative, long-term atorvastatin-treated group (ATL). At the end of the study, ATW-6 mice did not differ in any of the aforementioned parameters compared to their controls (CO-12W).
All atorvastatin groups showed reduced TNF-α and MCP-1 positive-stained areas in comparison to the relative controls. However, the reduction in MCP-1 was alleviated in ATW-3D group (24.00% ± 5.87%), since it showed significantly higher concentrations within plaques with respect to ATS group (13.14% ± 5.73%, P < .01).
Regarding MMP-2 concentrations, nonsignificant difference was found between all groups (P < .05). On the other hand, short-term discontinuation of atorvastatin resulted in higher concentrations for MMP-3 and MMP-9. In particular, MMP-3 plaque content in ATW-3D and ATW-7D groups was significantly higher than in ATS group (P < .05). The MMP-9 in ATW-3D also exhibited statistically significant higher concentrations (21.31% ± 4.87%) compared to ATS (12.35% ± 3.82%, P = .02) while ATW-7D showed no statistically significant increase when compared to the ATS (P > .05). Contrariwise, there was a significantly lower TIMP-2 concentration in ATW-3D group rather than ATS group (P < .05), while ATW-7D presented nonsignificant decrease in comparison with ATS (P > .05, Figures 4 and 5).
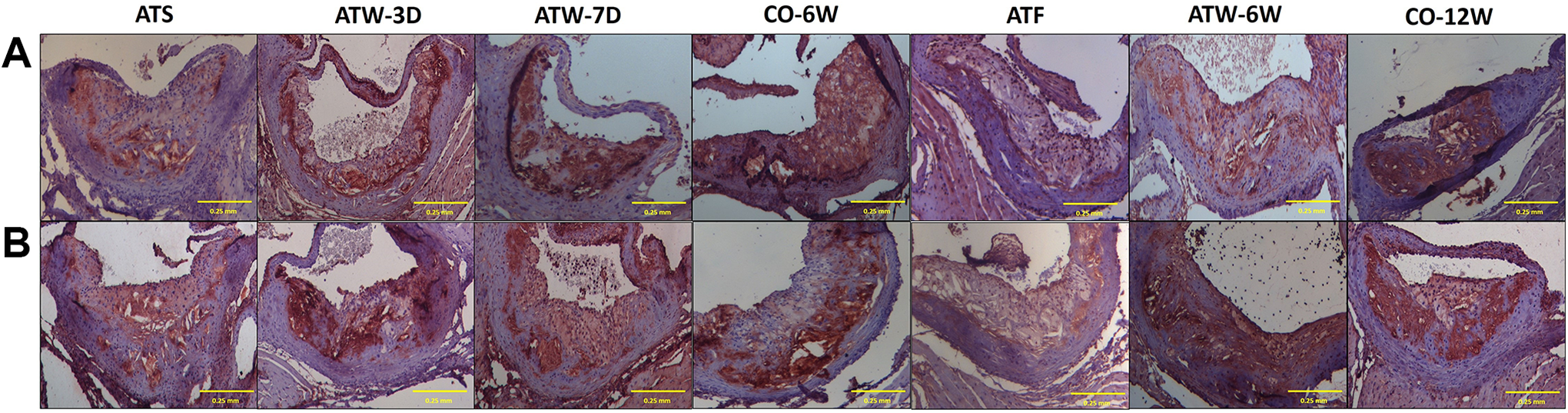
Immunohistochemical staining with antibodies against (A) macrophages (upper panel) and (B) MMP-9 (lower panel). Sections were counterstained with H&E (×20; scale bar: 0.25 mm).
Immunohistochemistry results in the examined region for antibodies against (A) a-actin, (B) mac-3, (C) MCP-1, (D) TNF-a, (E) MMP-2, (F) MMP-3, (G) MMP-9, and (H) TIMP-2. *Statistically significant difference compared to CO-6W. **Statistically significant difference compared to CO-12W. ***Statistically significant difference compared to ATS.
Statistically significant difference compared to both ATS and CO-6W.
Statistically significant difference compared to ATL.
Discussion
In the present experimental study, we assessed the impact of short-term and long-term withdrawal of atorvastatin on atherosclerosis progression and composition in ApoE−/− atherosclerotic mice. Acutely after cessation of atorvastatin administration, we demonstrated an abrupt atherosclerotic plaque destabilization. Thereafter, plaque stability was almost “restored” and after acute phase it started declining again in a less steep manner. A similar pattern of changes was observed for macrophage numbers and the majority of their inflammatory derivatives within plaques, including MCP-1, TNF-α, MMP-3, and MMP-9. Their concentrations were remarkably increased shortly after atorvastatin withdrawal, and that elevation persisted to a lesser extent as long as mice remained free from atorvastatin.
Statin-treated mice have shown increase collagen plaque contents. 23 –26 In a previous study from our team, ApoE−/− mice treated with atorvastatin 10 mg/kg exhibited decreased macrophages and increased collagen content compared to controls, while histological assessment did not reveal any significant differences in smooth muscle cells. 12 In agreement to those studies, we observed that the uninterrupted administration of atorvastatin, for both short and long time, considerably increased plaque collagen and connective tissue contents within plaques, while reduced lesion size and macrophages concentrations in the aorta of the same animal model. Beyond the significant changes in lipid levels in atorvastatin-treated mice throughout the study, we focused on atorvastatin’s nonlipid related, anti-inflammatory mechanisms. Thereby, continuous atorvastatin administration resulted in considerable suppression of the inflammatory milieu, as depicted by the lower contents of macrophages, MMP-3, MMP-9, MCP-1, and TNF-α. The key role of the latter factors in atherosclerotic plaque instability has been widely proved. 27 Accumulating data support the statin-induced inhibition of the inflammatory pathway involving MMP-9, MCP-1, and TNF-α, which potentially contributes to plaque stabilization. 28,29 Our results confirmed that both short- and long-term, uninterrupted statin therapy yielded in significant atherosclerosis regression and stabilization associated with the anti-inflammatory properties of statins. 30
Previous clinical studies have documented acute cardiovascular events early after cessation of statins’ uptake, without assessment of the concomitant alterations in atherosclerotic plaques composition. In large-scale registries, such as the National Registry of Myocardial Infarction 4 31 and the Global Registry of Acute Coronary Events (GRACE), 32 patients with ACS who discontinued statin rapidly lost statins’ benefits and experienced worse outcomes. 33 Those clinical results led previous investigators to postulate an acute destabilization of atherosclerotic plaques induced by statins’ withdrawal. To our knowledge, this is the first study demonstrating the abrupt reversion of the abovementioned antiatherosclerotic and stabilizing effects of a statin member shortly after its discontinuation (3 days). At that time point, the atherosclerotic plaque area remained unaltered; however, mice showed the lowest concentrations of collagen and connective tissue within plaques. That reversion was maintained but to a lesser extent thereafter (7 days and 6 weeks). Hence, the observed “rebound” phenomenon may provide a reasonable explanation for the aforementioned high incidence of cardiovascular events in clinical studies shortly after patients discontinue statin therapy.
Looking for a mechanistic explanation, we documented a nonsignificant change in cholesterol levels after atorvastatin discontinuation, indicating that other than lipid-lowering mechanisms are involved in acute plaque destabilization. Most importantly, we found an acute, shortly after atorvastatin’s withdrawal, remarkable increase in macrophages content within atherosclerotic lesions, equivalent to controls’ levels, and remained thereafter. We tested whether those changes in macrophage numbers could influence the production and secretion of macrophages derivatives, like MMPs and inflammatory mediators, which are associated with plaque vulnerability. With regard to this hypothesis, TNF-α and MCP-1 increased significantly and abruptly after atorvastatin removal. The TNF-α elevation persisted as long as atorvastatin administration was stopped. Notably, MCP-1 concentration peaked at 3 days and was “compromised” subsequently, implicating an acute inflammatory response. The MCP-1 firing may be related to nuclear factor κB (NF-κB) activation, resulting in the induction of pro-inflammatory cytokines, such as interleukin1b and TNF-α, 34,35 which exacerbate plaque development and vulnerability. 9,36 In parallel, TNF-α along with MCP-1 stimulate the expression and release of MMPs, such as MMP-3, playing an important role in fibrous cap thinning and atherosclerotic plaque disruption. 37 Data from human pathology studies have demonstrated an increase in MMP-3 levels in the “shoulder” regions of atherosclerotic plaques, where plaque rupture is usually observed. 38 Therefore, it is likely that the interruption of statin administration ceased suppressing the atherosclerotic inflammatory infiltration, leading to a remarkable release of MMP-3 production shortly after atorvastatin’s discontinuation (ATW-3D group). Another plausible explanation for MMP-3 elevation within plaques comes from the observed association between the number of macrophages, cellular carriers of MMP-3, and their derivative MMP-3.
In addition to MMP-3 levels, our analysis revealed a peak in MMP-9 levels in ATW-3D group. The MMP-9 has been extensively studied for its role in atherosclerosis, with numerous studies demonstrating its predominant role in the disease’s course by regulating the degradation of extracellular matrix (ECM). 39 The MMP-9 has an active presence throughout the atherogenesis, promoting plaque destabilization by macrophage infiltration and degradation of the fibrous cap. 40 The beneficial effect of statin administration on MMPs concentrations where shown in a study by Fulumoto et al 41 where MMP-3 and MMP-9 levels were lowered, within macrophage-containing areas in the aortas of hyperlipidemic rabbits. Besides this, histopathological evaluation of rabbits, mice, and human atherosclerotic samples has shown high concentration of MMP-3 and MMP-9 in areas with increased probability of rupture. 42
On the other hand, the observed acute decrease in TIMP-2, which counterbalances the detrimental effects of MMPs, indicates an imbalance in MMPs/TIMP ratio and the related ECM homeostasis within plaques. Therefore, the acute withdrawal of the suppressive effect of statins on inflammatory procedures, leads to a “rebound” stimulation of inflammatory factors resembling to “acute release of a coil.” At present, there are several articles that describe a rebound phenomenon of inflammatory response after statins withdrawal. 18,19 Our results indicate that statin discontinuation leads to inflammatory “explosion,” which after the acute phase seems to persist at even lesser degree. That inflammatory exaggeration could in turn induce acute plaque destabilization with potential clinical impact, such as myocardial ischemia, MI, and death.
Several limitations applied in the extrapolation of our results in clinical routine settings. The ApoE−/− mice is a valid animal model for the study of the atherosclerosis pathophysiology, since atherosclerotic plaques show similar characteristics to those in human beings. 20,21 However, there are differences with humans in cholesterol metabolism, lipid profile, and cardiovascular physiology. 43,44 Moreover, spontaneous plaque rupture is rare in mice, and for technical reasons we were unable to study coronary arteries. Instead, we followed a standardized protocol of our laboratory which allowed the colocalization for the various measured variables. Hence, the assessment of plaque vulnerability can be conducted indirectly via the composition of the plaque in ECM components and inflammation markers. 45 Finally, we did not measure the gene expression of inflammatory agents due to the huge amount of required analysis.
Conclusion
In conclusion, the uninterrupted atorvastatin administration attenuated atherosclerosis progression and enhanced plaque stability via the suppression of inflammatory potential within the aortic atherosclerotic lesions of hypercholesterolemic ApoE−/− mice. When atorvastatin therapy was suddenly ceased, its atheroprotective effect seemed to be revoked promptly, leading to acute plaque destabilization, independent of cholesterol levels changes. The acute release of inflammatory mediators within the atherosclerotic plaques after statin’s discontinuation may explain mechanistically the exaggerated and unfavorable alterations in plaque stability in the short-term period. After initial rebound response, those effects were slightly smoothed thereafter, leading eventually to a sustained plaque destabilization in the long term. Our findings are of clinical importance outlining the risk of adverse events in atherosclerotic patients who do not comply to continuous statins’ therapy.
Supplemental Material
supplemantary_file - Statins’ Withdrawal Induces Atherosclerotic Plaque Destabilization in Animal Model—A “Rebound” Stimulation of Inflammation
supplemantary_file for Statins’ Withdrawal Induces Atherosclerotic Plaque Destabilization in Animal Model—A “Rebound” Stimulation of Inflammation by Marianna Stasinopoulou, Nikolaos P. E. Kadoglou, Eirini Christodoulou, Efthymios Paronis, Nikolaos G. Kostomitsopoulos, Georgia Valsami, Christos D. Liapis and John Kakisis in Journal of Cardiovascular Pharmacology and Therapeutics
Footnotes
Acknowledgments
The authors would like to thank Pfizer Company for providing the pure atorvastatin substance.
Author Contribution
Stasinopoulou, M, Kadoglou, N, Kostomitsopoulos, N, Valsami, G, Liapis, C, and Kakisis, J contributed to conception or design. Stasinopoulou, M, Kadoglou, Christodoulou, E, and Paronis, E contributed to acquisition, analysis, or interpretation. Stasinopoulou, M and Paronis, E. Kadoglou, N, Kostomitsopoulos, N drafted the manuscript. Kostomitsopoulos, N, Valsami, G, Liapis, C, and Kakisis, J critically revised the manuscript. All authors gave final approval and agree to be accountable for all aspects of work ensuring integrity and accuracy.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.