
Abstract
Background:
Naloxegol is a novel selective, peripherally acting μ-opioid receptor antagonist for treating opioid-induced constipation (OIC) in patients with chronic pain syndromes. We analyzed the cardiovascular (CV) safety of naloxegol based on data from its development program prior to approval by the US Food and Drug Administration in 2015.
Methods:
Comprehensive CV safety analyses were performed in 4 clinical studies of naloxegol (12.5 and/or 25 mg) in patients with noncancer pain and OIC: two 12-week, double-blind, randomized studies; a 12-week, double-blind, extension study; and a 52-week, randomized, open-label study versus usual care. Evaluations of baseline CV risk were obtained from medical histories and clinical findings at the time of study initiation.
Results:
Across the 4 studies (N = 2135), 68% of patients had ≥1 CV risk factor and 41% had a history of CV disease, diabetes, or ≥2 other CV risk factors. There were no increases in blood pressure, heart rate, or the rate-pressure product with naloxegol versus placebo. The rates of major adverse cardiovascular events (MACE) per 100 patient-years of exposure were 1.13 (95% confidence interval [CI], 0.31-2.89) for placebo/usual care and 0.75 (95% CI, 0.24-1.75) for naloxegol. The relative risk of MACE for all doses of naloxegol versus placebo was 0.67 (95% CI, 0.14-3.36).
Conclusion:
These data demonstrate that naloxegol has a CV safety profile comparable to placebo/usual care in patients with OIC. Although the observed number of events was low, the data show no CV signal in patients with OIC treated with naloxegol.
Introduction
Opioid-induced constipation (OIC) is a common, burdensome side effect of long-term opioid therapy, affecting 40% to 80% of patients receiving chronic therapy. 1,2 Activation of μ-opioid receptors in the gastrointestinal tract leads to a decline in gastrointestinal motility and intestinal propulsion, increased water absorption from the bowel contents, and other alterations to bowel function, leading to OIC. 1 Peripherally acting μ-opioid receptor antagonists (PAMORAs) are a therapeutic class of agents used to treat OIC and postoperative ileus. 1,3 Peripherally acting μ-opioid receptor antagonists selectively block μ-opioid receptors in the gastrointestinal tract but do not cross the blood–brain barrier, preventing constipation while preserving analgesia. 1,3 Oral naloxegol was the first oral PAMORA approved for the treatment of OIC. Since the approval of naloxegol, 3 other PAMORAs have been approved: subcutaneous and oral methylnaltrexone for OIC, oral alvimopan for postoperative ileus, and naldemedine for OIC. 4 -7
Naloxegol is approved for the treatment of OIC in adult patients with chronic noncancer pain, 5 based on the results of several clinical studies of 12 to 52 weeks’ duration. 8 -10 Opioid receptor antagonists, including PAMORAs, could hypothetically cause rapid opioid withdrawal, resulting in increases in blood pressure (BP) and heart rate and a possible increased risk of cardiovascular (CV) events. 11 Based on this theoretical concern, CV safety was formally evaluated in the development program for naloxegol and included assessments of changes in vital signs and prospective adjudication of potential CV adverse events. 8 -10,12,13 We report the CV safety findings from the clinical development program for naloxegol in patients with noncancer pain and OIC.
Methods
These analyses included data from four phase 3 studies of oral naloxegol in patients with noncancer pain and OIC (Supplemental Table 1 and Figure 1). 8 -10 All studies were conducted in accordance with the Declaration of Helsinki and International Conference on Harmonisation/Good Clinical Practice guidelines, and all patients provided written informed consent. Study protocols and informed consent forms were approved by local independent ethics committees or institutional review boards. 8 -10
Study Designs
The studies used for this analysis are shown in Figure 1. In studies A and B, patients with documented OIC were randomized 1:1:1 to placebo, naloxegol 12.5 mg, or naloxegol 25 mg for 12 weeks. 8 In study C, patients who successfully completed study A continued on their randomized dose from the parent study. 10

Studies used in the cardiovascular safety analysis. aTotal number of patients randomized. bTotal number of patients enrolled.
In study D, patients were randomly assigned (2:1) to receive once-daily naloxegol 25 mg or usual care for OIC. Usual care was defined as a laxative treatment selected and prescribed by the investigator according to his/her usual clinical practice; no peripheral opioid antagonists or products containing naloxone were permitted as part of that treatment regimen. Throughout the study, laxative treatment could be adjusted at any time point for patients randomized to usual care. 9
Study Patients
Studies A and B included adult patients (aged 18-84 years) who were receiving stable oral opioid therapy (30-1000 morphine-equivalent units [MEUs] per day) for noncancer pain and had been on that regimen for ≥4 weeks. All eligible patients had active OIC, as indicated by <3 spontaneous bowel movements (BMs) per week and ≥1 key symptom of OIC (hard/lumpy stools, straining, or the sensation of incomplete evacuation or anorectal obstruction) present for ≥25% of BMs during the 4 weeks before screening. 8 Opioid-induced constipation was confirmed over 2 weeks based on patient-reported information related to BMs and laxative and opioid use. 8 Exclusion criteria, including CV history–related criteria, are described in the Supplemental Methods.
Study C included patients who had completed study A and were continuing to receive a stable maintenance opioid regimen (30-1000 MEUs). 10 Study D included patients who had completed study B or C; however, the majority (90%) of patients in study D were new patients (aged 18-85 years) with noncancer pain who had confirmed OIC and were receiving a stable opioid maintenance regimen (30-1000 MEUs). 9 The exclusion criteria used for these 2 studies were the same as those for studies A and B. 8
Assessments
All studies included comprehensive prospective evaluations of CV safety, including vital signs (seated BP and pulse). Timing for these evaluations is summarized in the Supplemental Methods. The heart rate–BP product (or rate-pressure product) was calculated as the heart rate × the systolic BP (beats/min·mm Hg). The procedure for evaluating CV outcomes, including adverse events and adjudicated major adverse cardiovascular events (MACE), is also summarized in the Supplemental Methods. Electrocardiography (ECG) data are presented for studies A, B, and D. In studies A and B, 12-lead ECGs were performed at screening, at each of the study visits during treatment, and at the final study visit. In study D, 12-lead ECGs were performed at screening, at 8 visits during treatment, and at the final study visit. On the day of the first dose, a pre- and postdose ECG was performed, with the postdose timing corresponding to the estimated peak drug concentration. All ECGs were centrally read by a cardiologist experienced in digital ECG and clinical trials.
A post hoc assessment of CV risk at baseline was performed by evaluating case report forms for the following risk factors: age ≥75 years, diagnosis of hypertension or taking an antihypertensive medication, diagnosis of hyperlipidemia or using a lipid-lowering medication, current smoker, use of low-dose aspirin for a CV-related indication, diagnosis of type 2 diabetes or taking insulin or oral antihyperglycemic agents, and a medical history of CV disease. 14,15
Statistical Analyses
For each of these studies, safety analyses were conducted for patients in the intent-to-treat population who had received ≥1 dose of study medication. For the purposes of this analysis, study A and its extension study, study C, were considered as 1 study and denoted as study A/C. Data from the placebo-controlled studies, including studies A/C and B, were pooled for these analyses because these studies had similar designs and treatment durations (Figure 1). Study D was considered separately for these analyses because the control arm and study duration differed from the placebo-controlled studies (Figure 1).
Data from all studies were pooled to allow for an overall assessment of the incidence of MACE for naloxegol and its comparator (placebo or usual care). For the pooled analysis, the number of events, patient-years of exposure, exposure-adjusted incidence rates, and relative risk (RR) for naloxegol versus placebo/usual care were assessed. Confidence intervals (CIs) for the incidence rates and RR were estimated using exact methods based on the Poisson distribution to account for the low number of events observed.
The number and percentage of patients with individual CV risk factors at baseline were tabulated overall and by treatment group. Changes from baseline in systolic BP, heart rate, and the rate-pressure product were evaluated at day 1 (at 1 hour post-treatment) and for the last on-treatment measure and were summarized by treatment group using descriptive statistics (mean and standard error). The numbers and percentages of patients with systolic BP increases and decreases of ≥20 mm Hg, 10 to 20 mm Hg, or 0 to 10 mm Hg were shown by treatment group. The numbers and percentages of patients with heart rate changes of ≥20 beats per minute (bpm), 15 to 20 bpm, 10 to 15 bpm, 5 to 10 bpm, and 0 to 5 bpm were also shown by treatment group. Electrocardiography data were pooled for studies A and B and evaluated separately for study D. Mean changes in ECG parameters from baseline were evaluated for all postbaseline measures. The number and percentage of patients with ECG parameter values outside of prespecified extended reference ranges (used to identify potentially clinically important ECG abnormalities) were tabulated by the treatment group.
These studies were not designed or powered to specifically evaluate CV effects, including MACE. Therefore, no formal statistical analyses were performed.
Results
Patients and Baseline CV Risk
Patient characteristics were generally similar across all treatment groups within each of the studies (Table 1). Patients in these studies were typically 65 years or younger and primarily women, and the majority of patients had ≥1 CV risk factor or disease. Over one-fourth of all patients (27.3% [582/2135]) had 1 CV risk factor at baseline, excluding diabetes or a history of CV disease, while 40.9% (874/2135) of patients had a history of CV disease, diabetes, or ≥2 other CV risk factors. Over half of all patients (52.2% [1115/2135]) were on a therapeutic agent for hypertension, hyperlipidemia, or CV disease prevention.
Characteristics and Cardiovascular Risk Factors at Baseline.
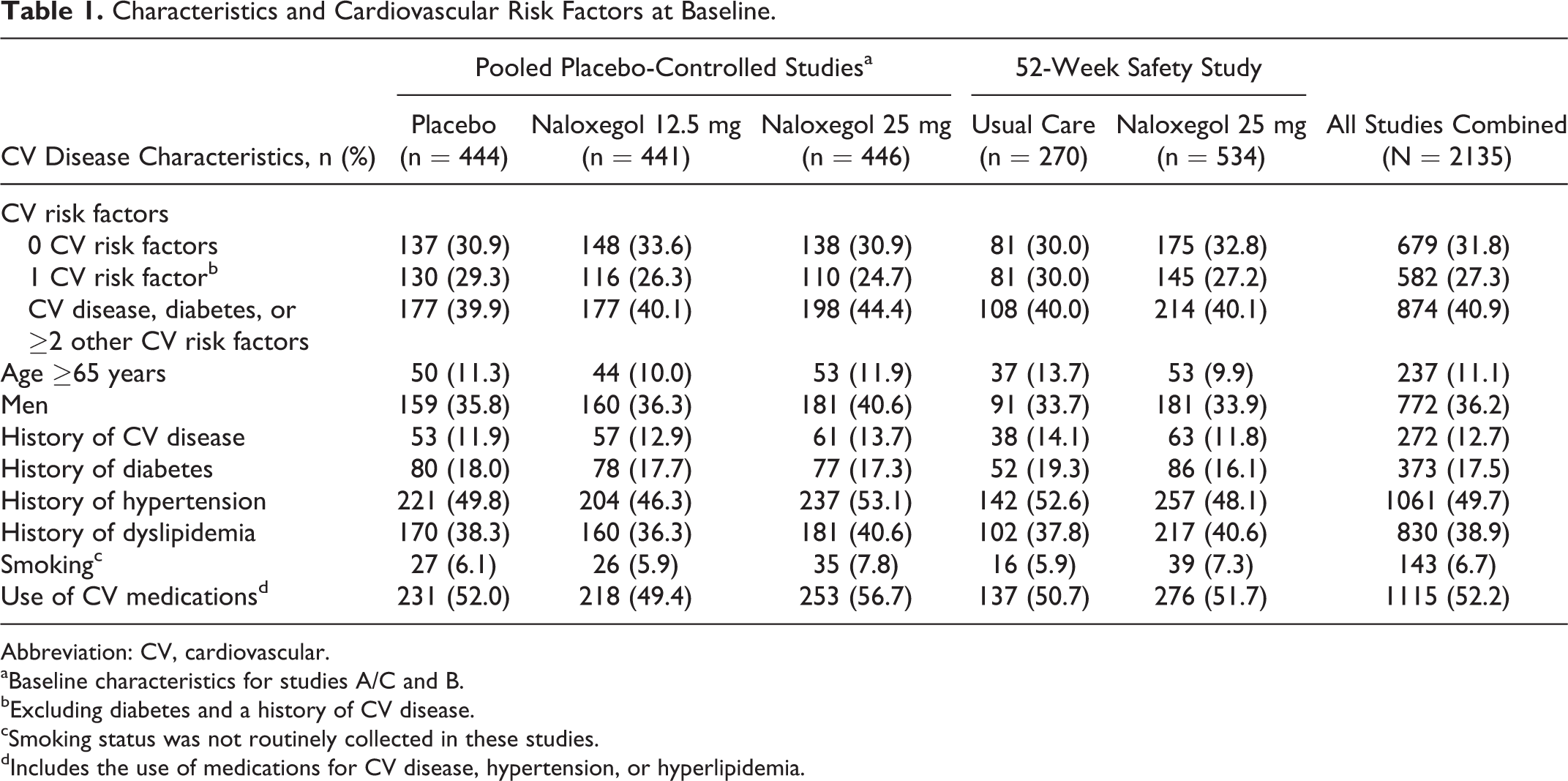
Abbreviation: CV, cardiovascular.
aBaseline characteristics for studies A/C and B.
bExcluding diabetes and a history of CV disease.
cSmoking status was not routinely collected in these studies.
dIncludes the use of medications for CV disease, hypertension, or hyperlipidemia.
Blood Pressure, Heart Rate, and Electrocardiograms
Mean changes from baseline in systolic BP at day 1 (1 hour after the first dose, coinciding with peak effect time of naloxegol) and the last on-treatment measurement were comparable in the placebo and naloxegol (12.5 and 25 mg) treatment groups in the pooled placebo-controlled studies; changes from baseline in systolic BP ranged from 0.7 to 1.6 mm Hg at 1 hour after the first dose and from 0.4 to 1.3 mm Hg at the last on-treatment measurement (Table 2). Mean changes from baseline in systolic BP at day 1 and at the last on-treatment measurement were also similar in the usual care and naloxegol 25 mg treatment groups of the 52-week safety study (Table 2).
Mean Changes From Baseline in Systolic BP, Heart Rate, and Rate-Pressure Product.a,b

Abbreviations: BP, blood pressure; SE, standard error.
aData are expressed as mean ± SE of the mean.
bNo imputation method was used for missing values.
cThe rate-pressure product was calculated as the heart rate × systolic BP.
Mean changes from baseline in heart rate were comparable across the placebo and naloxegol (12.5 and 25 mg) treatment groups in the placebo-controlled studies, with decreases of −2.4 to −2.7 bpm observed at 1 hour after the first dose of study treatment and increases of 1.0 to 1.8 bpm observed at the last on-treatment measurement (Table 2). The mean changes from baseline in heart rate were also low and similar for patients receiving usual care and those receiving naloxegol 25 mg in the 52-week safety study at 1 hour after the first dose of study treatment and the last on-treatment measurement (Table 2).
Mean changes from baseline to 1 hour after the first dose of study treatment and to the last on-treatment measurement in the rate-pressure product (heart rate × systolic BP) were similar in the placebo and naloxegol (12.5 and 25 mg) treatment groups in the placebo-controlled studies, with decreases ranging from approximately −177 to −236 bpm·mm Hg at 1 hour after the first dose and increases ranging from approximately 210 to 260 bpm·mm Hg at the last on-treatment measurement (Table 2). Change from baseline to 1 hour after the first dose of study treatment and to the last on-treatment measurement in the rate-pressure product were also similar in the usual care and naloxegol 25 mg treatment groups in the 52-week safety study (Table 2).
Results of analyses of the distribution of changes in BP and heart rate from baseline that were intended to identify patients with larger changes in systolic BP or heart rate from baseline to 1 hour after the first dose of study treatment on day 1 are shown in Figures 2 and 3, respectively. The proportion of patients with a decrease or increase in systolic BP of ≥20 mm Hg from baseline to 1 hour after the first dose of study treatment was low (≤2% and ≤6%, respectively) and similar across all treatment groups in the placebo-controlled studies and 52-week safety study (Figure 2), as was the proportion of patients with a decrease or increase in heart rate of ≥15 bpm in the placebo-controlled studies (≤3% and ≤1%, respectively; Figure 3).

Categories of changes in systolic BP from baseline on day 1 at 1 hour postdose in the (A) placebo-controlled studies (studies A/C and B) and (B) 52-week safety study (study D). BP denotes blood pressure.

Categories of changes in heart rate from baseline on day 1 at 1 hour postdose in the (A) placebo-controlled studies (studies A/C and B) and (B) 52-week safety study (study D). bpm denotes beats per minute.
No clinically important treatment differences were observed in either the mean changes from baseline in ECG parameters or in the proportion of patients with ECG parameter values outside of prespecified reference ranges. The number and proportion of patients with potentially clinically important ECG abnormalities, based on prespecified ranges, at 2 hours after the first dose of study drug and at any time during the treatment period are summarized in Supplemental Tables 2 and 3, respectively. Although a slight imbalance between treatment groups was observed in a number of potentially clinically important QRS duration abnormalities (≥140 milliseconds), individual review of these results showed that the majority of these patients either had an abnormal baseline QRS duration or a history of CV disease. Thus, the QRS duration results did not raise a safety concern for naloxegol.
Cardiovascular Adverse Events
Rates of serious adverse CV events (including events in the Medical Dictionary for Regulatory Activities [MedDRA] system organ classes [SOCs] of “cardiac disorders” or “vascular disorders”) observed during the treatment period are shown for the overall population and subgroups according to baseline CV risk in the placebo-controlled studies and for patients in the 52-week safety study in Table 3. Incidences of serious adverse CV events were low (<2% in all groups) and comparable across the placebo and naloxegol (12.5 and 25 mg) treatment groups in the placebo-controlled studies, as well as in the usual care and naloxegol 25 mg groups in the 52-week safety study. Incidences of serious adverse CV events were also low and similar across patient groups divided by the number of risk factors, with serious adverse CV event incidences ranging from no events in patients with no CV risk factors to 4 to 5 events per treatment group in those patients with multiple risk factors or CV disease at baseline (Table 3).
CV Serious Adverse Events Reported During the Treatment Period by CV Risk Category at Baseline and CV/Cerebrovascular Serious Adverse Events and Adjudicated MACE.

Abbreviations: CV, cardiovascular; MACE, major adverse cardiovascular event; MI, myocardial infarction; SAE, serious adverse event.
aAny CV SAE in the Medical Dictionary for Regulatory Activities (MedDRA) system organ classes (SOCs) of “cardiac disorders” or “vascular disorders.”
bExcluding diabetes and a history of CV disease.
cIncludes information gathered during the treatment period and follow-up.
dIncludes SAEs in the MedDRA cardiac SOC, vascular SOC, and cerebrovascular standardized MedDRA queries (SMQs).
eOne patient had a nonfatal MI but died later in the study.
Incidences of CV/cerebrovascular serious adverse events (including events in the MedDRA cardiac SOC, vascular SOC, and cerebrovascular standardized MedDRA query) and adjudicated MACE in the placebo-controlled studies (studies A/C and B) and 52-week safety study (study D) are summarized in Table 3. Overall, 10 events in 9 patients were adjudicated as MACE; the numbers and proportions of MACE were similar across treatment groups in both the placebo-controlled studies and the 52-week safety study (Table 3). Additionally, 1 patient who received naloxegol 25 mg in study C and 1 patient who received usual care in study D were each adjudicated as having been hospitalized due to heart failure.
In the placebo-controlled studies (studies A/C and B), there were 2 MACE in the placebo group (132.7 patient-years of exposure), 2 MACE in the naloxegol 12.5 mg group (128.9 patient-years of exposure), and 1 MACE in the naloxegol 25 mg group (126.0 patient-years of exposure). In the 52-week safety study, there were 2 MACE in the usual care group (222.2 patient-years of exposure) and 2 MACE in the naloxegol 25 mg group (411.0 patient-years of exposure). Across all studies, there were 4 MACE in the placebo/usual care group (354.9 patient-years of exposure) and 5 MACE in the naloxegol group (all doses; 665.9 patient-years of exposure). The rate of MACE per 100 patient-years of exposure was 1.13 (95% CI, 0.31-2.89) for placebo/usual care and 0.75 (95% CI, 0.24-1.75) for all doses of naloxegol across all studies. The RR of MACE for naloxegol compared to placebo/usual care was 0.67 (95% CI, 0.14-3.36).
Discussion
The principal findings of this comprehensive CV safety analysis show that naloxegol, an oral PAMORA, had comparable changes from baseline in BP, heart rate, and the rate-pressure product to placebo and usual care. An outlier analysis, which assessed the proportion of patients with increases in systolic BP of ≥20 mm Hg at the peak effect time of naloxegol, also showed comparable effects for naloxegol and placebo in the placebo-controlled studies and for naloxegol and usual care (ie, laxatives without naloxegol) in the 52-week safety study. There were also similarities in the rate-pressure product, a surrogate of myocardial oxygen demand, 16 between the placebo/usual care and naloxegol groups, which suggests that naloxegol does not meaningfully contribute to excessive cardiac demands. No clinically important treatment differences were observed in ECG parameters with naloxegol compared with placebo or usual treatment. Rates of MACE and other serious adverse CV events were low and similar in number to naloxegol and placebo/usual care.
Not surprisingly, the proportion of patients with serious adverse CV events was higher for those patients who had CV disease, diabetes, or ≥2 other CV risk factors at baseline compared with those with no or 1 CV risk factor; however, within these CV risk subgroups, the proportions of patients with serious adverse CV events were similar for naloxegol and placebo or usual care. Based on the RR and acknowledging the relatively small number of adjudicated CV events and duration of follow-up, there was no evidence of an imbalance in CV events with naloxegol versus placebo (or versus usual care).
Results of these analyses support those of the individual 52-week safety study 9 and the 12-week, placebo-controlled studies, 8,10 which showed a low incidence of MACE and no association of increased risk of CV events with naloxegol. Along with the findings of those studies, a thorough phase 1 QT study in 52 healthy male volunteers showed no clinically important changes in any ECG parameters, including heart rate, QT interval, PR interval, RR interval, and QRS duration, with naloxegol treatment at therapeutic (25 mg) and supratherapeutic (150 mg) doses. 13 Furthermore, these results provide additional support for the CV safety of agents in the PAMORA therapeutic class. 1,3,17 -21 In the development program of another PAMORA, alvimopan, there were reports in one 12-month safety study of increased rates of myocardial infarction for patients with OIC receiving alvimopan compared with those receiving placebo. 5 However, this unfavorable CV safety finding has not been observed uniformly in other studies of alvimopan. 18,19 In fact, in a retrospective, matched cohort study of alvimopan in patients undergoing bowel resection, the incidence of CV morbidity was significantly lower among patients receiving alvimopan (19.4%) than among matched patients who were not treated with alvimopan (24.0%; P = .0001). 18 In a separate pooled analysis of phase 3 studies of alvimopan (6 and 12 mg) for postoperative ileus following bowel resection, incidences of hypertension, hypotension, and tachycardia were comparable in the placebo and alvimopan treatment groups. 19 Similarly, neutral CV safety findings have been observed with the PAMORA methylnaltrexone; incidences of tachycardia and hypotension were numerically higher with placebo (both 6%) than with methylnaltrexone (2% and 0%, respectively). 21 Our results are also comparable to those for patients receiving the PAMORA naldemedine in 2 pivotal phase 3 studies, where <1% of patients experienced a major CV event. 22
Our safety analysis of naloxegol is subject to certain limitations. As with other development programs for drugs not intended for the treatment of CV diseases, the studies, while large in sample size, were not powered to definitively rule out small or intermediate differences in CV events between treatment groups. Hence, the relatively low number of CV events observed across these studies results in low precision around estimates or incidence rates and RRs and precludes a more robust statistical assessment of treatment differences. Although no clinically important CV safety risks were identified based on these study data, an ongoing, postmarketing, observational, epidemiological study of naloxegol will provide additional data on CV safety. In the current study, the overall lack of imbalance across treatment groups, along with the comparable changes in BP, heart rate, and rate-pressure product, indicates that naloxegol has a CV safety profile comparable to that of placebo and/or usual care.
Supplemental Material
Supplemental Material, JCPT_Oct_2016_225_revised_CV_effect_revised_supplemental_materials_24_Jan_2018 - Cardiovascular Safety of the Selective μ-Opioid Receptor Antagonist Naloxegol: A Novel Therapy for Opioid-Induced Constipation
Supplemental Material, JCPT_Oct_2016_225_revised_CV_effect_revised_supplemental_materials_24_Jan_2018 for Cardiovascular Safety of the Selective μ-Opioid Receptor Antagonist Naloxegol: A Novel Therapy for Opioid-Induced Constipation by William B. White, Peter Kowey, Ulysses Diva, Mark Sostek, and Raj Tummala in Journal of Cardiovascular Pharmacology and Therapeutics
Footnotes
Acknowledgments
The authors thank Megan Knagge, PhD, of MedErgy (Yardley, Pennsylvania) for providing editorial assistance, which was in accordance with Good Publication Practice (GPP3) guidelines and funded by AstraZeneca (Wilmington, Delaware).
Author Contributions
All authors qualify for authorship. Dr William B. White drafted the manuscript and performed analyses. Dr Peter Kowey critically reviewed the manuscript and provided edits. Dr Ulysses Diva performed statistical testing of the data and critically reviewed the manuscript. Dr Mark Kostek was involved with study conduct and oversight and critically reviewed the manuscript. Dr Raj Tummula was involved with medical monitoring of the study and critically reviewed the manuscript.
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: No author received payment for manuscript production or editorial work. William B. White is a safety consultant to and receives consulting fees from AstraZeneca Pharmaceuticals. Peter Kowey is a paid consultant to AstraZeneca Pharmaceuticals. Ulysses Diva was an employee of AstraZeneca Pharmaceuticals LP when the study was performed and is currently affiliated with Ardea Biosciences, a subsidiary of AstraZeneca; he held and continues to hold stock from AstraZeneca. Mark Sostek was an employee of AstraZeneca Pharmaceuticals LP and held stock options from AstraZeneca when the study was performed. Raj Tummala is an employee and shareholder of AstraZeneca Pharmaceuticals LP.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Supplemental Material
Supplementary material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.