
Abstract
Myocardial infarction (MI) is a serious cardiovascular disease resulting in high rates of morbidity and mortality. Although advances have been made in restoring myocardial perfusion in ischemic areas, decreases in cardiomyocyte death and infarct size are still limited, attributing to myocardial ischemia/reperfusion (I/R) injury. It is necessary to develop therapies to restrict myocardial I/R injury and protect cardiomyocytes against further damage after MI. Many studies have suggested that peroxisome proliferator-activated receptor γ (PPARγ), a ligand-inducible nuclear receptor that predominantly regulates glucose and lipid metabolism, is a promising therapeutic target for ameliorating myocardial I/R injury. Thus, this review focuses on the role of PPARγ in cardioprotection during myocardial I/R. The cardioprotective effects of PPARγ, including attenuating oxidative stress, inhibiting inflammatory responses, improving glucose and lipid metabolism, and antagonizing apoptosis, are described. Additionally, the underlying mechanisms of cardioprotective effects of PPARγ, such as regulating the expression of target genes, influencing other transcription factors, and modulating kinase signaling pathways, are further discussed.
Keywords
Introduction
Myocardial infarction, one of the most serious cardiovascular diseases, causes a serious threat to human health and results in high rates of morbidity and mortality. 1 Although it is important to salvage viable myocardium by emergent restoration of myocardial perfusion in ischemic areas, reperfusion itself can trigger complex pathophysiological processes and even exacerbate the injury initially induced by ischemia, termed myocardial ischemia/reperfusion (I/R) injury. 2 Reperfusion injury has been shown to extend the infarct size and cause myocardial dysfunction, such as myocardial stunning, arrhythmias, heart failure, and even cardiac arrest. 3,4
Fortunately, recent studies involving myocardial I/R injury and cardiac protection show encouraging developments. Peroxisome proliferator-activated receptor γ (PPARγ), a ligand-inducible nuclear receptor belonging to the nuclear transcription factor superfamily, 5 is a representative example. Accumulating studies demonstrate that PPARγ can protect cardiomyocytes against I/R injury through regulating cardiac oxidative stress, 6 –8 inflammatory responses, 9 –11 glucose and lipid metabolism, 12 –14 and apoptosis. 15 –17 With respect to the underlying mechanisms, PPARγ plays an essential role in regulating gene expression (eg, glucose transporter-4 [GLUT-4], 12,13 adiponectin, 18 and nuclear factor κ B inhibitor α [IκBα] 19 ), influencing transcription factors (eg, nuclear factor κ B [NF-κB] 20 ), and modulating kinase signaling pathways (eg, protein kinase B [AKT], 12,21 c-Jun N-terminal kinase [JNK], 15 and extracellular signal-regulated kinase [ERK] 22 ).
In this review, briefly pathophysiological mechanisms of myocardial I/R injury, especially those influenced by PPARγ, will be firstly described. Subsequently, characteristics, ligands, and general function of PPARγ will be discussed to show an overview of PPARγ. Most importantly, cardioprotective effects of PPARγ against I/R injury and underlying PPARγ-mediated mechanisms will be discussed in detail.
Mechanisms of Myocardial I/R Injury
Since myocardial I/R injury is one of the important obstacles to decrease cardiomyocyte death and infarct size after restored myocardial perfusion, 23 mechanisms of myocardial I/R injury have been studied and elucidated in detail. In this respect, excessive oxidative stress is regarded as one of the primary causes of myocardial I/R injury. 24 When oxidases (eg, inducible nitric oxide [NO] synthase and nicotinamide adenine dinucleotide phosphate [NADPH] oxidase) are uncontrollably activated accompanied by mitochondrial dysfunction, 25 –27 reactive oxygen species (ROS; eg, NO and hydrogen peroxide [H2O2]) are generated in excess, enhancing oxidative stress during reperfusion. 24
Additionally, oxidative stress is also increased associated with abnormal myocardial inflammatory responses, 28 which is another important reason contributing to myocardial I/R injury. 29 In pathophysiological progression of abnormal myocardial inflammatory responses, NF-κB signaling pathway plays an essential role in promoting the expression of multiple pro-inflammatory factors and leukocyte adhesion molecules. 10,30,31 Along with these factors, individually or interactively, inflammatory cells infiltrate in reperfusion areas, thereby increasing tissue vulnerability and causing further damage. 10,32 Additionally, infiltration of inflammatory cells also leads to endothelial dysfunction, which is accompanied by decreased expression and coupling of endothelial NO synthase (eNOS), subsequently aggravating myocardial I/R injury. 33,34
Cardiac metabolic dysfunction of glucose and lipid, which is associated with dysfunction of GLUT-4 expression and translocation, is also an important reason, exacerbating myocardial I/R injury. 13,35 On the one hand, dysfunction of glucose uptake and utilization reduces production of adenosine triphosphate, thereby exacerbating cardiomyocyte energy crisis. 13 On the other hand, as fatty acid (FA) oxidation relies on normal glucose metabolism, 36 dysfunction of glucose metabolism results in abnormal lipid metabolism, which promotes ROS generation and contributes to deleterious effects on cardiomyocytes. 36 –38 In addition to metabolic dysfunction, cardiomyocyte apoptosis, which is associated with quantitative disorders of apoptosis-related proteins 39,40 and mitochondria dysfunction induced by calcium paradox, 41,42 also aggravates myocardial I/R injury.
An Overview of PPARγ
Peroxisome Proliferator-Activated Receptor γ as a Nuclear Receptor
Peroxisome proliferator-activated receptor γ is an isoform of PPARs, which are ligand-inducible nuclear receptors belonging to the nuclear transcription factor superfamily. The other 2 isoforms of PPARs are PPARα and PPARβ/δ. 43 With respect to the distributions of PPARs, PPARα is predominantly expressed in liver, heart, kidney cortex, and skeletal muscle. 44 Peroxisome proliferator-activated receptor β/δ is ubiquitous, 45 whereas PPARγ is predominantly expressed in adipose tissue and macrophages, as well as in hepatocytes, cardiomyocytes, kidney, and skeletal muscle. 46
Evidence from previous studies has suggested that there are 2 detected proteins, PPARγ1 and PPARγ2, which are principally encoded by 4 messenger RNAs (mRNAs) including PPARγ1, PPARγ2, PPARγ3, and PPARγ4. 47 The mRNAs of PPARγ1, PPARγ3, and PPARγ4 encode the same PPARγ1 proteins, 48,49 whereas the mRNA of PPARγ2 encodes PPARγ2 proteins. 48 With respect to PPARγ transcription, PPARγ1 mRNA is found in almost all tissues at varying levels, while PPARγ2 mRNA is predominantly found in adipose tissue. 50 Peroxisome proliferator-activated receptor γ3 mRNA is distributed in macrophages, adipose tissue, and T lymphocyte, whereas PPARγ4 distribution has not been fully elucidated. 49,51
Under diseased conditions, the expression of PPARγ in heart changes to some degree. It has been observed that decreased cardiac expression of PPARγ is associated with diet-induced obesity, 52 while the expression of PPARγ is increased accompanied by decreased expression of PPARα in cardiomyocytes under diabetes mellitus. 53 Moreover, after myocardial ischemia, the expression of PPARγ is upregulated, whereas the expression of PPARα and PPARβ/δ is not altered in the infarcted areas. 54
Peroxisome proliferator-activated receptor γ is activated by ligands to execute its biological effects. These effects, involving oxidative stress, inflammatory responses, glucose and lipid metabolism, and cellular apoptosis, are summarized in Table 1 and will be hereinafter described in detail.
Effects of PPARγ Against Myocardial I/R Injury.
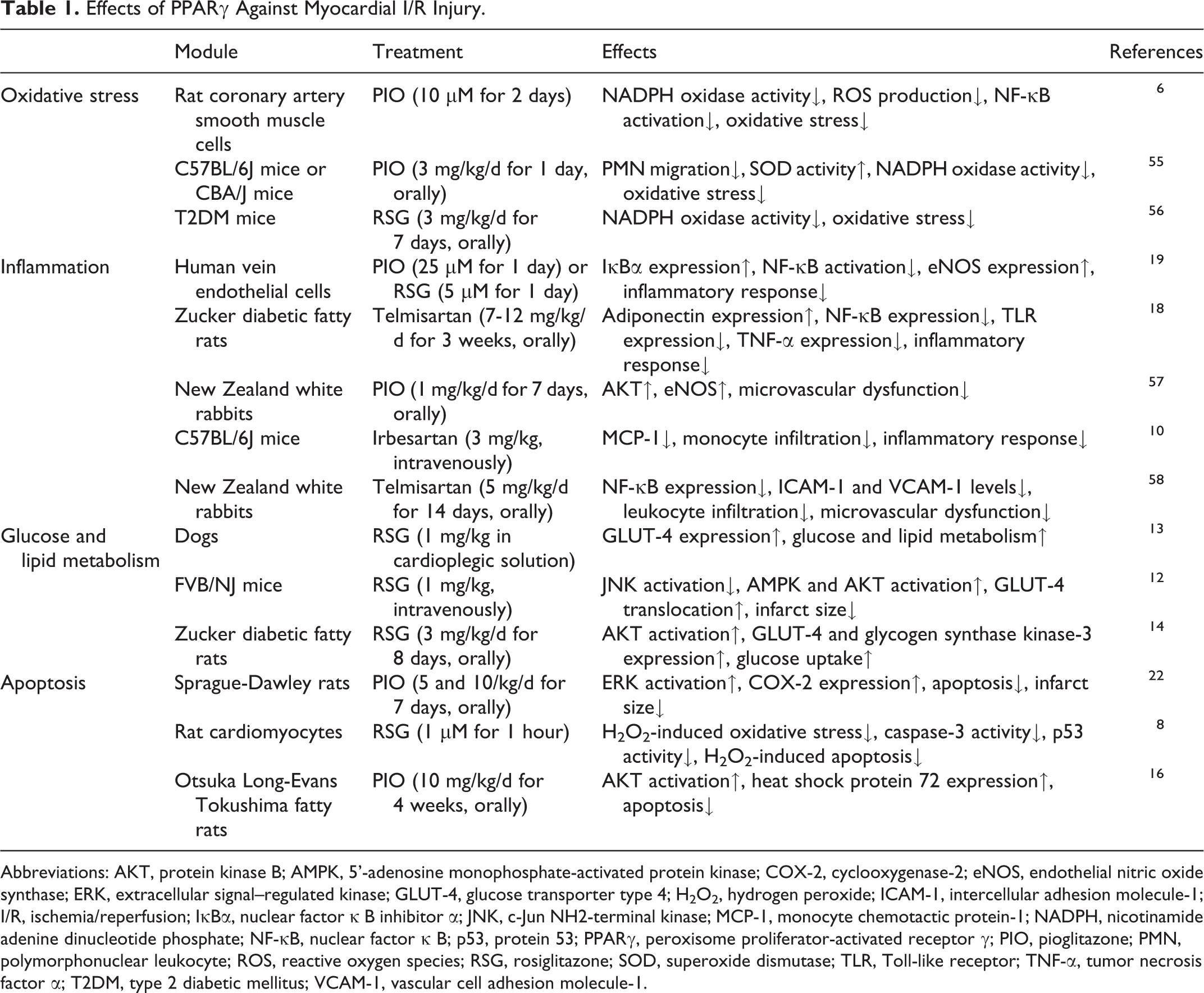
Abbreviations: AKT, protein kinase B; AMPK, 5’-adenosine monophosphate-activated protein kinase; COX-2, cyclooxygenase-2; eNOS, endothelial nitric oxide synthase; ERK, extracellular signal–regulated kinase; GLUT-4, glucose transporter type 4; H2O2, hydrogen peroxide; ICAM-1, intercellular adhesion molecule-1; I/R, ischemia/reperfusion; IκBα, nuclear factor κ B inhibitor α; JNK, c-Jun NH2-terminal kinase; MCP-1, monocyte chemotactic protein-1; NADPH, nicotinamide adenine dinucleotide phosphate; NF-κB, nuclear factor κ B; p53, protein 53; PPARγ, peroxisome proliferator-activated receptor γ; PIO, pioglitazone; PMN, polymorphonuclear leukocyte; ROS, reactive oxygen species; RSG, rosiglitazone; SOD, superoxide dismutase; TLR, Toll-like receptor; TNF-α, tumor necrosis factor α; T2DM, type 2 diabetic mellitus; VCAM-1, vascular cell adhesion molecule-1.
The Ligands of PPARγ
The ligands of PPARγ are divided into endogenous ligands and synthetic ligands. 59 Endogenous ligands principally include FA metabolites and their derivatives such as eicosanoids (eg, prostaglandins, 12-, and 15-hydroxyeicosatetraenoic acid) and conjugated linoleic acids (eg, 9- and 13-hydroxyoctadecadienoic acid). 59,60 Particularly, a prostaglandin, 15-deoxy-Δ 12,14 -prostaglandin J2 (15d-PGJ2) is regarded as a representative endogenous ligand of PPARγ. Interestingly, the level of 15d-PGJ2 in cardiac tissues is increased after myocardial I/R, which is related to the activation of cyclooxygenase 2 (COX-2). 11
As for synthetic ligands, thiazolidinediones (TZDs), well-known antidiabetic drugs, are typical synthetic ligands of PPARγ. 44 Rosiglitazone (RSG) and pioglitazone (PIO) are 2 representative TZDs, which are commonly used and have potent effects on activating PPARγ, whereas other TZDs, including ciglitazone and troglitazone, are lesser used. 44,61 Moreover, several angiotensin receptor blockers, including telmisartan 62 and irbesartan, 63 are demonstrated to be partial agonists of PPARγ receptors, while dual PPARs agonists including dual PPARα/γ agonists (eg, aleglitazar 21 and GCP-02 64 ), dual PPARδ/γ agonists (eg, compound 23 and compound 20 65 ), and dual PPARα/δ agonists (eg, T0913659 66 ) can correspondingly activate 2 isoforms of PPARs. 66 Furthermore, there are emerging pan PPARs agonists (eg, bezafibrate 67,68 and chiglitazar 69 ), simultaneously activating all 3 isoforms and resulting in combined effects of PPARs.
Notably, although plenty of PPARγ ligands induce their responses by binding with PPARγ, some PPARγ ligands, including 15d-PGJ2 and TZDs, can also induce their biological effects through additional PPARγ-independent mechanisms. 8,11,70 These mechanisms, such as directly modulating kinase activity 70 and regulating gene transcription, 8,11 will be discussed in following sections.
Transcriptional Transactivation of PPARγ
Upon binding with endogenous or synthetic ligands, PPARγ is activated and translocates into the nucleus, thereby interacting with the retinoid X receptor (RXR) and forming a heterodimeric complex. The resulting PPARγ–RXR complex subsequently recruits coactivators, such as PPARγ coactivator-1α 5 , and binds to peroxisome proliferator-responsive elements (PPREs). 5,71 As the result of it, the productions encoded by PPARγ target genes, including those involving in lipid store, glucose metabolism (eg, adiponectin 18 and GLUT-4 13 ), oxidative stress (eg, manganese superoxide dismutase 72 ), and inflammatory responses (eg, IκBα 19 ), are significantly increased, which subsequently contributes to modulating cells survival. 71
Transcriptional Transrepression of PPARγ
In addition to transactivation, the activated PPARγ with endogenous or synthetic ligands can also negatively modulate relevant gene expression by directly inhibiting other transcription factors or competing with other transcription factors for coactivators, termed transcriptional transrepression, which is independent from PPREs. 5,73
A representative example is the NF-κB signaling pathway mediated by PPARγ. When NF-κB signaling pathway is resting, NF-κB is a tetramer including subunit p50, subunit p65, IκBα, and IκBβ. 31 When IκBα and IκBβ are disassociated from the tetramer associated with stimulation of pro-inflammatory factors, 74 the p50/p65 dimer is activated, recruiting their coactivators and subsequently forming a new complex. The new complex can bind to NF-κB-specific promoter sequences and induce the expression of other inflammatory factors (eg, tumor necrosis factor α, interleukin-1 [IL-1], interleukin-6 [IL-6], intercellular adhesion molecule-1 [ICAM-1], and vascular cell adhesion molecule-1 [VCAM-1]), which can promote inflammatory responses. 58,75,76
Activated PPARγ with endogenous or synthetic ligands can not only directly combine with the p50/p65 dimer and form a new complex that downregulates the NF-κB signaling pathway 20,77 but also upregulate the expression of IκBα and indirectly inhibit the transcriptional activity of NF-κb. 19 Additionally, the activator protein-1 signaling pathway and the Janus kinase/signal transducer and activator of transcription signaling pathway are also inhibited by activated PPARγ through similar ways mentioned above. 5,78
Complementary Effects and Interactions Between PPARs
Combined activation of PPARs is believed to contribute to complementary and synergistic effects, especially those on energy metabolism. 66 Primarily, PPARα promotes FA uptake and β-oxidation, 79 whereas PPARγ promotes glucose metabolism, FA uptake, and storage but lightly FA oxidation. 80 Peroxisome proliferator-activated receptor β/δ, which is an emerging isoform of PPARs, contributes to improving FA β-oxidation and glucose oxidation. 81 Since all 3 isoforms of PPARs involve in modulating glucose and lipid metabolism, combined activation of PPARs can induce not only more effective promotion but also better counterbalance on energy metabolism. 64,66,68,82
Apart from metabolic effects, PPARs further exhibit nonmetabolic effects on anti-inflammation and antioxidation. Specially, PPARγ and PPARα can synergistically interact with the NF-κB signaling pathway and contribute to combined repression on inflammatory responses. 20,83 Besides, combined effects of PPARs on mediating kinase activity (eg, AKT, 21 5’-adenosine monophosphate-activated protein kinase [AMPK] 84 ) also play important roles in mediating inflammation and ameliorating I/R injury. 21,84
Additionally, interactions between PPARγ and other 2 PPAR isoforms have caused extensive concern. Since PPARβ/δ is a special PPAR isoform, which recruits corepressors and represses PPREs-mediated transcription when bound to DNA, PPARβ/δ has been shown to contribute to inhibiting PPARγ-mediated transcription. 85 Moreover, a similar suppressive effect of PPARβ/δ is also attributed to the competition for RXR between PPARγ and PPARβ/δ, which inhibits the form of PPARγ–RXR complex. 86 In contrast, pharmacological activation of PPARα has been shown to associate with promotive PPARγ-mediated transcription through upregulating the expression of PPARγ coactivator-1α. 87 These interactions between PPARγ and other PPAR isoforms allow for fine tuning of PPARγ signaling pathway and contribute to harmonizing efficacy and side effects of PPARγ. 88
Peroxisome Proliferator-Activated Receptor γ in Cardioprotection
Peroxisome Proliferator-Activated Receptor γ and Oxidative Stress
As discussed above, excessive oxidative stress, which is principally attributable to excessive ROS generation, is a key reason of aggravated myocardial I/R injury. 24 –26 Inhibition of ROS production potentially alleviates oxidative stress during myocardial reperfusion. In fact, accumulating studies have indicated that the production of ROS can be inhibited through PPARγ-mediated mechanisms. For instance, a recent study demonstrated that PIO (10 μM for 2 days) protected coronary artery smooth muscle cells against excessive oxidative stress by suppressing NADPH oxidase and ROS generation through a PPARγ-dependent mechanism. 6 Interestingly, a similar outcome was observed in murine heterotopic cardiac allotransplantation models, which pretreated with PIO (3 mg/kg/d for 1 day, orally, in mice) before cardiac transplantation and had less graft oxidant stress due to PPARγ-mediated suppression on NADPH oxidase. 55 Similarly, antioxidant effects of RSG in cardiac tissues have been observed. Kim et al reported that detrimental effects of H2O2-induced oxidative stress were limited through activation of PPARγ with RSG (1 μM for 1 hour, in rat cardiomyocytes). 8 Meanwhile, RSG upregulated thioredoxin expression in rat cardiomyocytes through an additional PPARγ-independent mechanism, thereby suppressing intracellular ROS generation and resulting in a combined effect on decreasing oxidative stress. 8 Through a PPARγ-independent mechanism, RSG (20 μM for 2 days, in human endothelial cells) directly activated AMPK and subsequently inhibited NADPH oxidase. 70 More importantly, apart from its antioxidant effects in healthy models, activated PPARγ by RSG (3 mg/kg/d for 7 days, orally) has further been shown to protect coronary arteriolar against NADPH oxidase-induced oxidative stress in type 2 diabetic mice. 56
Notably, antioxidant effects of activated PPARγ are further reported in some human trials. A clinical trial involving in 306 patients who underwent coronary artery bypass grafting has suggested that activated PPARγ upregulated adiponectin gene expression and subsequently decreased NADPH oxidase-mediated oxidative stress in heart tissue. 7 Interestingly, in this clinical trial, the activation of PPARγ was attributed to oxidation-induced 4-hydroxynonenal, a precursor of 13-hydroxyoctadecadienoic acid, 89 suggesting that physiological activation of PPARγ also protects heart tissue against excessive oxidative stress during I/R. 7
Peroxisome Proliferator-Activated Receptor γ and Inflammatory Responses
Since the NF-κB signaling pathway is generally recognized as one of the key mechanisms of myocardial inflammatory responses during I/R, 31 studies exploring the relationship between PPARγ and NF-κB have been performed. A recent study revealed the pivotal and irreplaceable role of PPARγ in inhibition on NF-κB activity through demonstrating that PPARγ-knockout mice had more increased NF-κB activity and more enhanced inflammatory responses in cardiomyocytes, compared with wild mice. 76
In contrast, activated by ciglitazone (5 mg/kg, intraperitoneally, in rats), PPARγ has been reported to suppress NF-κB activity, 11 which ameliorated myocardial inflammatory responses (Figure 1). 18,11 Through a similar PPARγ/NF-κB pathway, irbesartan (3.0 mg/kg, intravenously, in mice) has resulted in increased inhibition on monocyte chemotactic protein-1 (MCP-1) and attenuated accumulation of inflammatory monocytes in risky reperfusion areas. 10 More importantly, in a human trial involving in 319 patients with type 2 diabetic mellitus (T2DM), treatment with PIO (15 mg) has contributed to less myocardial I/R injury, possibly attributed to the suppression on inflammatory responses. 9

The underlying molecular mechanisms of PPARγ in cardioprotection. Upon binding with endogenous or synthetic ligands, PPARγ is activated. Activation of PPARγ attenuates the activity of JNK, thereby suppressing apoptosis by upregulating BCL-2 and suppressing caspase-3. In contrary, activated PPARγ enhances the activity of ERK, AMPK, and AKT. Activated ERK improves the activity of COX-2 and suppresses the expression of BAD and BAX, subsequently promoting antiapoptotic effects. As AMPK is activated in both PPARγ-dependent and PPARγ-independent manners, the expression of Glut-4 is upregulated contributing to ameliorated glucose metabolism, while the activity of NADPH oxidase is inhibited, resulting in attenuated oxidative stress. Associated with activated AKT, eNOS activity is enhanced and results in increased bioavailability of NO and ameliorated microvascular dysfunction, while the expression of GLUT-4 and HSP-72 is upregulated, respectively, contributing to ameliorated glucose metabolism and suppressed apoptosis. Furthermore, activated PPARγ can directly inhibits NF-κB and subsequently downregulates the expression of pro-inflammation cytokines (eg, IL-1, IL-6, and MPC-1) and leukocyte adhesion molecules (eg, ICAM-1 and VCAM-1), contributing to decreased inflammatory responses and ameliorated microvascular dysfunction. Upon binding with RXR, activated PPARγ can upregulate the expression of target genes, including IκBα and adiponectin. IκBα has been shown to inhibit NF-κB activity, whereas induced adiponectin can inhibit excessive cytosolic Ca2+, which promotes the mitochondria-dependent apoptosis and downregulates the expression of TLR, subsequently inhibiting TNF-α which can promote apoptosis and upregulate the expression of pro-inflammation cytokines including IL-1 and IL-6. Moreover, associated with upregulated adiponectin, AMPK activity is enhanced, whereas the activity of NADPH oxidase is suppressed and subsequently results in attenuated oxidative stress. AMPK indicates 5’-adenosine monophosphate-activated protein kinase; AKT, protein kinase B; BAD, BCL-2/BCL-XL-associated death promoter; BAX, BCL-2 associated X protein; BCL-2, B cell leukemia/lymphoma 2; COX-2, cyclooxygenase-2; eNOS, endothelial nitric oxide synthase; ERK, extracellular signal-regulated kinase; GLUT-4, glucose transporter-4; HSP-72, heat shock protein 72; ICAM-1, intercellular adhesion molecule-1; IL, interleukin; IκBα, nuclear factor κ B inhibitor α; JNK, c-Jun N-terminal kinase; MCP-1, monocyte chemotactic protein-1; NADPH, nicotinamide adenine dinucleotide phosphate; NF-κB, nuclear factor κ B; NO, nitric oxide; PPARγ, peroxisome proliferator-activated receptor γ; RXR, retinoid X receptor; TNF-α, tumor necrosis factor α; TLR, Toll-like receptor; VCAM-1, vascular cell adhesion molecule-1.
Furthermore, adiponectin is increasingly regarded as a mediator between PPARγ and inflammatory responses because of its ability on modulating inflammatory signaling pathways. Induced by PPARγ with telmisartan (7-12 mg/kg/d for 3 weeks, orally, in diabetic rats), adiponectin downregulated the expression of Toll-like receptors, thereby suppressing the expression of TNF-α and NF-κB. 18 Suppressed TNF-α contributes to downregulation of the expression of other inflammatory cytokines including IL-1 and IL-6, 90 while inhibited NF-κB is responsible for downregulating the expression of MCP-1, IL-1, and IL-6, both resulting in attenuated inflammatory responses (Figure 1). 10,76
Numerous reports suggest that cardiac microvascular dysfunction, which is associated with inflammatory responses during I/R, 91 is limited through PPARγ/adiponectin-mediated mechanisms. 34,57,58 As shown by Margaritis et al, PPARγ-induced adiponectin restored eNOS coupling in microvascular endothelial cells, which ameliorated the dysfunction of NO generation and release, and subsequently ameliorated microvascular dysfunction (Figure 1). 34 Similarly, PIO (1 mg/kg/d for 7 days, orally, in rabbits) has been shown to drive the PPARγ/AKT pathway and subsequently contribute to the restoration of eNOS in cardiac tissues after I/R. 57 Moreover, it was reported that telmisartan (5 mg/kg/d for 14 days, orally, in rabbits) downregulated the expression of leukocyte adhesion molecules including ICAM-1 and VCAM-1 in cardiac microvascular endothelial cells through a PPARγ/NF-κB-dependent mechanism (Figure 1). 58 Attributed to decreased expression of leukocyte adhesion molecules, leukocyte adhesion and infiltration are attenuated, subsequently contributing to ameliorative microvascular dysfunction and decreased inflammatory responses during myocardial I/R. 58,91
Notably, aleglitazar (150-600 nM for 1 day, in human cardiomyocytes), a dual PPARα/γ agonist, has shown additive effects on improving microvascular dysfunction and suppressing apoptosis, attributed to the stimulations of both PPARα/AKT/eNOS and PPARγ/AKT/eNOS signaling pathways. 21 Similarly, after treatment with bezafibrate (400 mg/kg/d for 21 days, orally, in type I diabetic rats), a pan PPAR agonist, myocardial angiogenesis was improved, which was attributed to combined activation of PPARs and responsible for protecting heart against ischemia injury. 67
Peroxisome Proliferator-Activated Receptor γ and Cardiac Glucose and Lipid Metabolism
Since decreased expression of GLUT-4 in cardiomyocyte has been shown to contribute to aggravated cardiac glucose and lipid metabolic dysfunction during I/R, 35 enhanced GLUT-4 expression possibly improves cardiac metabolism and subsequently ameliorates myocardial I/R injury. 92 –94 In fact, Zhang and Tang, who investigated small interfering RNA-mediated PPARγ gene knockdown in rats, demonstrated that cardiac metabolic dysfunction of glucose and lipid could be induced by PPARγ deficiency. 95 Conversely, another recent study has indicated that acute treatment with RSG (1 mg/kg) increased the expression of GLUT-4 and enhanced glucose uptake and utilization in dog hearts through a PPARγ-dependent mechanism. 13 Interestingly, the similar effect of acute treatment with RSG (1 mg/kg, intravenously, in mice) on increased expression of GLUT-4 was also associated with both PPARγ/AMPK and PPARγ/AKT pathways (Figure 1). 12 Consequently, the increased expression of GLUT-4 in heart improves myocardial glucose metabolism, which is further responsible for ameliorated energy crisis, decreased abnormal lipid metabolism production, suppressed ROS generation, and inhibited apoptosis, subsequently attenuating myocardial I/R injury. 37,38,94,96
Moreover, similar beneficial metabolic effects of PPARγ during myocardial I/R have further been demonstrated in both diabetic animal models and humans. As shown by Yue et al, short-term treatment with RSG (3 mg/kg/d for 8 days, orally) contributed to increased expression of glycogen synthase kinase-3 and GLUT-4 and subsequently improved cardiac glucose uptake and utilization in diabetic rats through a PPARγ/AKT pathway. 14 Similarly, in a human trial involving in 51 patients with T2DM and coronary artery disease, short-term treatment with RSG (4-8 mg, for 16 weeks) has shown to decrease circulating lactate concentrations, which indicates the improvement of myocardial glucose and lipid uptake and utilization. 97 These improvements of myocardial energy metabolism consequently contribute to better cardiomyocyte survival with less reperfusion injury.
However, activation of PPARγ by synthetic ligands, especially long-term activation, has shown to contribute to excessive lipid accumulation in cardiomyocyte, attributed to the promotion of PPARγ on FA uptake and triglyceride storage, but weakly promotion on utilization of FA and triglyceride. 53,98 Subsequently, because of the imbalance between lipid storage and utilization, abnormal lipid intermediates accumulate and lead to cardiomyocyte dysfunction or death, which diminishes the beneficial metabolic effects of PPARγ 99 and at least in part contributes to some cardiac negative outcomes (eg, cardiac hypertrophy) of treatment with some PPARγ synthetic ligands. 53,99,100
Notably, since activated PPARα is elementary in stimulating lipid consumption through enhancing FA oxidation, the dual or pan agonists of PPARs theoretically harmonize lipid store effects and beneficial metabolic effects of PPARγ in cardiac tissues. 64,66 In fact, in a study involving diet-induced diabetic rats, treatment with bezafibrate (30 mg/kg/d for 6-8 weeks, orally) has been demonstrated to result in both decreased triglycerides store and improved glucose metabolism in cardiac tissues, attributed to its combined effects on activating both PPARα and PPARγ. 68 Furthermore, bezafibrate lowered the 30-day rehospitalization rates and crude 1-year mortality of patients after acute coronary syndromes, supporting the effects of pan PPAR agonists against myocardial reperfusion injury. 82
Thus, it is thought that short-term treatment with PPARγ agonists is beneficial to improve cardiac glucose and lipid metabolism during I/R, whereas long-term treatment with PPARγ agonists potentially contributes to imbalanced cardiac lipid metabolism and diminishes the beneficial cardioprotective effects of PPARγ. Through combined activation of PPARs, emerging pan PPAR agonists are promising to harmonize cardioprotective effects and related metabolic disorders of activation of PPARγ.
Peroxisome Proliferator-Activated Receptor γ and Cardiac Apoptosis
During myocardial reperfusion, the pivotal role of apoptosis in myocardial I/R injury has been frequently observed. Accumulating evidence demonstrate that myocardial I/R injury is limited by inhibiting cardiomyocyte apoptosis through PPARγ-dependent mechanisms.
On one hand, perfusion with RSG (1 μM) in isolated rat hearts before I/R has been reported to activate PPARγ, which subsequently inhibited JNK activity. 15 As activated JNK can aggravate apoptosis by inhibiting B cell leukemia/lymphoma 2 and stimulating caspase-3, the inhibition of JNK attenuates cardiomyocyte apoptosis (Figure 1). 101,102 Additionally, activation of PPARγ with PIO (10 mg/kg/d for 4 weeks, orally, in rats) further resulted in upregulated expression of heat shock protein 72, which protected cardiomyocyte against caspase-dependent apoptosis, through the PPARγ/AKT pathway (Figure 1). 16 On the other hand, PPARγ-induced adiponectin has been shown to attenuate excessive cytosolic Ca2+ and inhibit the subsequent mitochondria-dependent apoptosis by enhancing uptake of cytosolic Ca2+ into the sarcoplasmic reticulum in a sphingosine 1-phosphate/calmodulin kinase II signaling-dependent manner (Figure 1). 103 Furthermore, a recent study has demonstrated that antiapoptotic effects of PIO (5 and 10/kg/d for 7 days, orally, in rats) were associated with PPARγ-mediated enhanced activation of ERK, 22 which attenuated cardiomyocyte apoptosis and myocardial infarct size through not only suppressing pro-apoptotic proteins (eg, B cell leukemia/lymphoma 2 [BCL-2]/BCL-XL-associated death promoter and BCL-2-associated X protein) but also stimulating the COX-2/prostaglandin E2 pathway (Figure 1). 17,22
More interestingly, the natural PPARγ ligands, 15d-PGJ2 (0.5 mg/kg, intraperitoneally, in rats) was further found to result in antiapoptosis effects through promoting heat shock factor 1 binding to target genes that encoded heat shock proteins 70, 11 whereas PIO (10 mg/kg/d for 3 days, intraperitoneally, in PPARγ-knockout mice) contributed to anti-apoptosis effects through activating AKT and increasing expression of COX-2. 104 These observations suggest that antiapoptosis effects of PPARγ ligands can rely on additional PPARγ-independent mechanisms.
Failures of PPARγ Ligands in Cardioprotection
Notably, in contrast to general cardioprotective effects described above, PPARγ ligands have paradoxically failed to attenuate myocardial I/R injury in some studies. As shown by Xu et al, treatment with RSG (3 mg/kg/d for 8 weeks, orally, in pigs) has led to no significant effect on attenuating myocardial I/R injury, which was hypothetically attributed to the less severity of myocardial ischemia and suggested that the cardioprotective effects of RSG possibly associated with high severity of myocardial ischemia. 105
Even, possibly through a PPARγ-independent mechanism, acute preperfusion with high-dose troglitazone (at a mean myocardial concentration of 7.5 μg/g, in pigs) resulted in no attenuated myocardial I/R injury but increased susceptibility of cardiomyocyte to ventricular fibrillation, which was associated with ion channel dysfunction. 106 Similarly, another experiment, which was performed in isolated mice hearts and contained preperfusion with RSG (10 and 30 μM for 24 hours), led to enhanced oxidative stress and mitochondrial dysfunction after I/R, attributed to PPARγ-independent suppression on superoxide dismutase and inhibition on complexes I and IV in mitochondria. 107 Those results suggest that some PPARγ-independent pathways, at least in part, contribute to failures of PPARγ ligands in cardioprotection. It is possible that the higher risk of frustrated cardioprotection is associated with high dosage or concentrations of PPARγ ligands, which drive unexpected signaling pathways against cardioprotective effects of PPARγ.
Conclusion
As studies exploring the role of PPARγ in myocardial I/R injury have accumulated, a wide array of biological functions of PPARγ in cardioprotection during I/R have emerged. These cardioprotective effects of PPARγ, including attenuating oxidative stress, inhibiting inflammatory responses, improving glucose and lipid metabolism, and antagonizing apoptosis, suggest that PPARγ is a promising therapeutic target for ameliorating myocardial I/R injury. The mechanisms underlying these pleiotropic effects form a complex network involving regulation on target gene expression, influence on other transcription factors, and modulation on kinase signaling pathways, which interactively decrease cardiomyocyte damage in I/R.
However, although cardioprotective effects of PPARγ are demonstrated in plenty of experimental studies and human trails, regrettable failures of treatment with PPARγ synthetic ligands in cardioprotection have been reported, which are possibly associated with some PPARγ-independent pathways or adverse side effects of PPARγ activation. In order to prevent these negative effects of PPARγ synthetic ligands, further studies probing into more detailed elucidation of both PPARγ-dependent and PPARγ-independent mechanisms are required. Advances in more detailed clarification of molecular mechanisms will promote to develop PPARγ-dependent treatment strategies for alleviating myocardial I/R injury.
Footnotes
Author Contributions
Chong-Bin Zhong, Xi Chen, and Xu-Yue Zhou contributed equally to this article.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was funded by the National Natural Science Foundation of China (No: 81400190).