
Abstract
Myocardial ischemia/reperfusion injury represents a major threat to human health and contributes to adverse cardiovascular outcomes worldwide. Despite the identification of numerous molecular mechanisms, understanding of the complex pathophysiology of this clinical syndrome remains incomplete. Thioredoxin-interacting protein (Txnip) has been of great interest in the past decade since it has been reported to be a critical regulator in human diseases with several important cellular functions. Thioredoxin-interacting protein binds to and inhibits thioredoxin, a redox protein that neutralizes reactive oxygen species (ROS), and through its interaction with thioredoxin, Txnip sensitizes cardiomyocytes to ROS-induced apoptosis. Interestingly, evidence from recent studies also suggests that some of the effects of Txnip may be unrelated to changes in thioredoxin activity. These pleiotropic effects of Txnip are mediated by interactions with other signaling molecules, such as nod-like receptor pyrin domain-containing 3 inflammasome and glucose transporter 1. Indeed, Txnip has been implicated in the regulation of inflammatory response and glucose homeostasis during myocardial ischemia/reperfusion injury. This review attempts to make the case that in addition to interacting with thioredoxin, Txnip contributes to some of the pathological consequences of myocardial ischemia and infarction through endogenous signals in multiple molecular mechanisms.
Introduction
Ischemia is a condition characterized by an acute or chronic decrease in blood flow to tissue. The primary consequence caused by ischemia is insufficient delivery of oxygen and nutrients, which in turn induces bioenergetic failure. Ischemia causes a profound metabolic stress that leads to serious tissue damage. Due to the nature of high metabolic demand, both the brain and the heart are organs particularly susceptible to ischemic insults. Although the mechanisms of ischemia/reperfusion injury in these vital organs are complicated, current proposed theories include oxidative stress, metabolic derangements, and inflammation—all of which lead to apoptotic or necrotic cell death.
In recent years, a number of studies suggest the involvement and the importance of thioredoxin-interacting protein (Txnip) in diverse biological processes and pathological conditions, including ischemia/reperfusion injury. 1 -8 Thioredoxin-interacting protein, also known as Vitamin D3–upregulated protein 1 or thioredoxin-binding protein 2, belongs to a group of proteins called α-arrestins. Thioredoxin-interacting protein, which is widely expressed in multiple cellular compartments, 9 has been shown to regulate a variety of biological processes ranging from oxidative metabolism to programmed cell death. As its name suggests, one of the most defining features of Txnip is its ability to bind to cytosolic and mitochondrial thioredoxins. 10,11 Moreover, Txnip is the only arrestin domain–containing protein 12 that interacts with thioredoxin, an ubiquitous antioxidant that contributes to the maintenance of the reducing environment in the cell. Two major isoforms of thioredoxin have been identified in mammalian cells: cytoplasmic thioredoxin-1 and mitochondrial thioredoxin-2. 13 In each isoform, the reduced form of thioredoxin serves a key component of the major redox system by functioning as an active scavenger of thiol-oxidized proteins to detoxify reactive species in the cell. The interaction between Txnip and thioredoxin is mediated by disulfide bonding at the redox-active catalytic site of thioredoxin. 14 As a negative regulator of thioredoxin and its reducing capacities,9 Txnip inhibits the reducing activity of thioredoxin 14 and its antioxidant effects through disulfide formation with thioredoxin. As such, Txnip acts as a pro-oxidant and induces apoptosis. 15
Thioredoxin-interacting protein is known to have pleiotropic functions. 16 In addition to its ability to inhibit thioredoxin, Txnip has important roles in glucose metabolism. 17 High glucose levels strongly induce Txnip messenger RNA (mRNA) expression in many cell types, 18,19 as well as in diabetic patients 20 through a well-defined transcriptional mechanism that is activated by an increase in intracellular glucose-6-phosphate. 21,22 Thioredoxin-interacting protein, in turn, blocks glucose transport into the cell, thereby returning Txnip levels toward normal as cellular glucose level decreases. Although it was initially proposed that Txnip affects glucose metabolism by inhibiting thioredoxin activity and increasing reactive oxygen species (ROS), Txnip actually retains its ability to control glucose metabolism even when it is unable to interact with thioredoxin. 23 Recent studies demonstrate that Txnip inhibits glucose transport through direct interaction with glucose transporter 1 (GLUT1). 24,25 Degradation of Txnip on energy crisis via an energy sensor-mediated mechanism results in an acute increase in GLUT1 function, thus enhancing glucose uptake.24 Given the fact that Txnip is a potent inhibitor of glucose transport and a negative regulator of aerobic glycolysis,1 Txnip gene is considered to be a tumor suppressor gene. In fact, the expression of Txnip is found to be downregulated in many cancer cell types, and low expression of Txnip correlates with poor clinical outcomes in several cancers. 26,27 The loss of Txnip leads to many features of glycolytic phenotypes and Warburg effects in cancer cells, 28 which is considered to be an adaptive response to low-oxygen environments within the tumors.
Hence, ample evidence now demonstrates that Txnip is a potent modulator of oxidative metabolism and several metabolic pathways. These changes in cellular redox balance and energy metabolism can largely translate into functional changes in the cardiovascular system, particularly in pathways that are associated with hypoxia/oxidation or ischemia/reperfusion injury. In this review, we summarize the current knowledge on the role of Txnip in ischemia/reperfusion injury.
The Expression Level of Txnip and Ischemic Stress
Although Txnip expression is upregulated by glucose19 and vitamin D3, 29 it is suppressed by diverse stimuli including oxidative stress, growth factors, and mechanical strain in cardiomyocytes. 15,30 Because the same stimuli often do not alter thioredoxin expression levels, stimuli that do change Txnip expression may comprise a critical mechanism that regulates the level of “available” thioredoxin capable of influencing downstream signaling pathways under pathological conditions. 15,30 For some stimuli such as glucose, the transcriptional mechanisms that induce Txnip expression have been well documented. However, it remains unclear whether other stimuli, in particular ischemic stress, specifically change Txnip gene expression.
Thioredoxin-interacting protein is shown to be a hypoxia-induced gene in many cell types. 31,32 Mouse hearts subjected to hypobaric hypoxia in decompression chamber show an increase in Txnip gene expression. 33 In contrast, another group suggests that hypoxia induces a rapid decrease in Txnip mRNA and protein expression in a hypoxia-inducible factor-independent manner in HEK293 and Hela cells. 34 A recent report further suggests that Txnip mRNA expression is regulated in a biphasic manner by hypoxia, whereby Txnip expression exhibits an initial rapid decrease followed by an increase under prolonged hypoxia. This biphasic effect of hypoxia on Txnip expression is mediated via the inhibition of downstream signaling of the mechanistic target of rapamycin complex 1 (mTORC1). 35 Based on these findings, it appears that Txnip expression can be either downregulated or upregulated by ischemic stress, depending on the cell type and the specific pathological condition. However, it remains unclear whether the effect of reperfusion itself is distinct from the effect of hypoxia or ischemia alone. Despite these unknown factors, the high sensitivity of Txnip expression to a number of different stimuli suggests that Txnip is a molecular switch that responds to diverse cellular stresses and regulates several molecular mechanisms in ischemia/reperfusion injury.
Thioredoxin-Interacting Protein Exacerbates Ischemia/Reperfusion Injury
Regardless of the controversy over the changes in the expression level of Txnip by ischemia, there seems to be considerable agreement on the effects of Txnip in ischemia/reperfusion injury. A number of animal studies indicate that the functions of Txnip during ischemia/reperfusion are harmful. A knockdown of Txnip in cardiomyocytes significantly reduces apoptosis and enhances cell survival under conditions of oxidative stress in vitro.2 Direct intracardiac injection of the DNA enzyme that reduces myocardial Txnip mRNA expression at the time of acute myocardial infarction leads to prolonged reduction in cardiomyocyte apoptosis in vivo, which in turn is associated with significant improvements in cardiac function.2 Moreover, Txnip acts as a mediator of ischemia/reperfusion injury in the heart under diabetic conditions. Thioredoxin-interacting protein RNAi decreases ischemia/reperfusion-induced oxidative stress and apoptosis in cardiomyocytes treated with high concentration of glucose.5 As evidenced by decreased myocardial infarct size and caspase-3 activity, myocardial injury in Txnip small interfering RNA (siRNA)-treated rat hearts with hyperglycemia is markedly alleviated compared to that of control hearts.5 Collectively, these data demonstrate a direct role of Txnip in the adverse effects of ischemic and oxidative stresses on cardiomyocyte survival and cardiac function.
Serious tissue damage mediated by Txnip under ischemic conditions has been reported in noncardiac organs as well. In the brain, the expression level of Txnip is increased in the cytoplasm of neurons with significant brain injury from focal cerebral ischemia in mice.4 A knockdown of Txnip using Txnip siRNA protects neuronal cells, increases cell viability, and decreases lactate dehydrogenase release following oxygen–glucose deprivation and reoxygenation.4 In the liver, in a rat model of hepatic ischemia/reperfusion injury, postischemic treatment with trans-resveratrol decreases Txnip expression with an associated increase in the reducing activity of thioredoxin.3 In the vasculature, a knockdown of Txnip was able to restore blood flow and rescue diabetes-related impairment of ischemia-mediated angiogenesis in diabetic mice with surgical introduction of hind limb ischemia. 36 Together, these findings support the important roles of Txnip in multiple organ damage induced by ischemia/reperfusion.
Oxidative Stress and Txnip-Mediated Ischemic Damage
As described, Txnip has pleiotropic functions. 16 Thioredoxin-interacting protein–mediated ischemia/reperfusion injury is reported to involve multiple mechanisms, including enhancement of ROS production, disorder of energy homeostasis, and activation of apoptotic signaling. Among them, the most well-known function of Txnip is its role as a pro-oxidant by inhibiting the reducing activity of thioredoxin. 13,14 An in vitro analysis demonstrates that adenoviral gene transfer of Txnip results in a significant increase in cellular levels of superoxide and apoptosis in cardiomyocytes following hypoxia/reoxygenation injury.5 The reciprocal effects have been demonstrated in the same system: overexpression of thioredoxin or knockdown of Txnip strongly inhibits the increase in superoxide and apoptosis by hypoxia/reoxygenation injury.5 It is well established that the increased production of ROS causes oxidative stress, which is one of the major mediators that damage multiple cellular components including nucleic acids, proteins, and lipids. Reactive oxygen species can stimulate transcription factors such as nuclear factor kappa B and activator protein-1 via mitogen-activated protein kinases, which induce apoptotic program in cardiomyocytes through signaling pathways. 37,38 In addition, oxidative stress damages organelles such as the endoplasmic reticulum and mitochondria, which facilitate the release of Ca2+ ions and proapoptotic proteins into the cytosol to enhance the apoptotic pathways. 39 Thioredoxin-interacting protein mediates these biological processes and promotes oxidative damage to tissues via its pro-oxidative effects. 30,40
However, it should be noted that in vivo data from two independently generated knockout mice model show that deletion of Txnip results in little change in cytosolic thioredoxin-1 activity. 17,41 These data suggest a possibility that although forced overexpression of Txnip can inhibit the activation of cytosolic thioredoxin-1, physiological levels of Txnip may not be sufficient to suppress its activity. 42 This can be explained by the observation that Txnip primarily exists in the nuclei and mitochondria, 43 indicating that the Txnip–thioredoxin interaction is not normally localized in the cytosol. Indeed, under physiological conditions, Txnip is primarily located in the nucleus. However, oxidative stress shuttles Txnip into mitochondria, where Txnip binds to and inhibits mitochondrial thioredoxin-2.11 In this respect, mitochondrial thioredoxin-2 may be more relevant to Txnip-mediated ischemia/reperfusion injury than cytosolic thioredoxin-1 (Figure 1).

Possible mechanism by which thioredoxin-interacting protein (Txnip) mediates ischemia/reperfusion (I/R) injury by interacting with mitochondrial thioredoxin-2 (TRX2). Ischemia/reperfusion induces cellular production of reactive oxygen species (ROS). In response to oxidative stress, Txnip is shuttled from the nucleus to mitochondria, where TRX2 regulates the generation of mitochondrial ROS and the mitochondrial apoptosis signaling pathway. Thioredoxin-2 also repairs reversible oxidative protein damage by reducing disulfide bonds that are formed by the oxidation of cysteines. Thioredoxin-interacting protein binds to TRX2 through a disulfide bond formation, inhibiting the reducing functions of TRX2. The interaction between Txnip and TRX2 promotes the generation of mitochondrial ROS and triggers the mitochondrial apoptosis signaling pathway, which leads to I/R injury.
Mitochondrial thioredoxin-2 regulates the rate of H2O2 emission flux from mitochondria, which is essential for keeping a redox environment that is compatible with the energetics of the heart. 44 Mitochondrial thioredoxin-2 controls programmed cell death by inhibiting the release of cytochrome c from the mitochondria and increasing the membrane potential of the mitochondria. 45 -47 Accordingly, the inhibitory effects of Txnip on mitochondrial thioredoxin-2 lead to oxidative stress and apoptosis in cardiomyocytes. We have previously found that mitochondrial thioredoxin-2 activity is decreased by myocardial ischemia/reperfusion via a direct interaction with Txnip.1 Following myocardial ischemia/reperfusion, Txnip knockout hearts in mice have a higher activity of mitochondrial thioredoxin-2, which is associated with lower ROS generation and better left ventricular mechanical function, compared to that of wild-type control hearts.1 A deficiency in Txnip confers a strong resistance to ROS-induced damage in a manner similar to the effects exhibited by an overexpression of mitochondrial thioredoxin-2. 48 Hence, the redox-sensitive interaction between Txnip and mitochondrial thioredoxin-2 is a crucial step in promoting myocardial ischemia/reperfusion injury.
Thioredoxin-Interacting Protein Links Oxidative Stress with Inflammatory Response in Ischemic Signaling
In addition to promoting pro-oxidative effects, recent studies highlight another role of Txnip as a key regulator that links oxidative stress to inflammation in the ischemic signaling pathway. In the pathogenesis of ischemia/reperfusion injury, ROS can work in concert with the inflammatory response. Ischemia-mediated generation of ROS is important for the activation of inflammatory signals in the myocardium. Reactive oxygen species induce cytokine expression, promote leukocyte integrin activation, and induce adhesion molecule synthesis. Thus, the inflammatory responses do play an important role in the overall pathogenesis of ischemic insults. Although the inflammatory process is necessary for tissue healing after myocardial infarction, it can also damage the myocardium and lead to maladaptive ventricular remodeling. 49
The initiation of inflammatory responses requires sensors, such as inflammasomes, to detect irregularities within the cellular components. Inflammasomes are inflammatory regulators that are recently identified to be multiprotein complexes that are essential for the initiation of many inflammatory responses in various cell types. The activation of inflammatory signaling pathways is required for the full inflammasome activation and mature cytokine production. There are four inflammasomes that have been identified thus far: nucleotide-binding oligomerization domain nod-like receptor pyrin domain-containing (NLRP) 1, NLRP3, nod-like receptor C4, and the protein absent in melanoma 2. 50 The NLRP3 inflammasome is a multiprotein complex consisting of NLRP3, apoptosis-associated speck-like protein containing a CARD (caspase recruitment domain) (ASC), and caspase-1 (Figure 2). The NLRP3 activates caspase-1 by interacting with ASC. The ASC bridges the interaction between NLRP and caspase-1, thereby contributing to inflammasome activation and the subsequent maturation and secretion of proinflammatory cytokine interleukin (IL) 1β and IL-18. 51 The formation of the NLRP3 inflammasome amplifies the inflammatory response and mediates tissue damage, leading to lipid metabolism disorders 52 and other human diseases. 53

The model of nucleotide-binding domain nod-like receptor protein 3 (NLRP3) inflammasome activation through reactive oxygen species (ROS) by ischemia/reperfusion (I/R). The I/R-induced ROS dissociate thioredoxin-interacting protein (Txnip) from cytosolic thioredoxin-1 (TRX1). Free Txnip then binds to NLRP3 and activates NLRP3 inflammasome. The NLRP3 oligomerizes and recruits apoptosis-associated speck-like protein containing a CARD (caspase recruitment domain) (ASC) and pro-caspase 1, triggering the activation of caspase 1. This biological process leads to the maturation of proinflammatory cytokine interleukin 1β (IL-1β). Accumulation of these cytokines within the myocardium induces I/R injury and contributes to cardiac dysfunction.
Interestingly, compelling evidence now suggests that Txnip binds to NLRP3 and subsequently triggers the activation of NLRP3 inflammasome. 54 In response to oxidative stress, Txnip dissociates from the complex containing thioredoxin and rapidly binds to NLRP3. Thioredoxin-interacting protein then induces inflammasomes activation, which results in the maturation and secretion of IL-1β and IL-18. 55 Thus, ROS-induced inflammatory response is regulated by Txnip, which triggers the assembly and oligomerization of inflammasomes under oxidative stress. 56 Considering the redox-sensitive features of Txnip-NLRP3 axis, it is reasonable to hypothesize that the interaction between Txnip and NLRP3 can be important in the development of tissue damage induced by ischemia/reperfusion. 57 The importance of this mechanism is underscored by several animal studies. First, a report suggests that the expression of NLRP3 is upregulated in myocardial infarction during the acute phase, which mediates myocardial ischemia/reperfusion injury. 58 Second, NLRP3 knockout mice are found to have better cardiac contractile function and smaller infarct size following ischemia/reperfusion injury than those of the control mice.58 Third, ASC knockout mice have reduced myocardial ischemia/reperfusion injury. 59 Fourth, IL-1β is a prominent mediator of inflammation in myocardial ischemia/reperfusion injury, whereby myocardial ischemia/reperfusion induces the expression of IL-1β, which when inhibited prevents myocardial injury due to ischemia/reperfusion. 60 Finally, cardiac NLRP3 inflammasome activation driven by Txnip modulates both the JAK2-STAT3 and insulin signaling pathways, leading to metabolic disorders in the ischemic heart. 61
However, the induced expression of NLRP3 by myocardial ischemic injury is primarily localized in noncardiomyocytes such as leukocytes, fibroblasts, and endothelial cells within the myocardium. 58,62 Cardiomyocytes, on the other hand, express a very low abundance of NLRP3 protein. 58 Even though the expression level of NLRP3 is limited in cardiomyocytes, NLRP3 is abundantly expressed in microvascular endothelial cells. 58,62 Ischemic stress is sufficient to increase the interaction between Txnip and NLRP3 in microvascular endothelial cells in the heart, which then induces an inflammatory response in the myocardium. Conversely, a ROS scavenger that dissociates Txnip from NLRP3 inhibits the activation of NLRP3 inflammasome in microvascular endothelial cells. Thus, interventions that block Txnip/NLRP3 interaction have the potential to confer therapeutic advantage in mitigating ischemia/reperfusion injury. This exciting possibility, in fact, has been tested in a recent study, which shows that intramyocardial Txnip siRNA injection decreases the endothelial interaction between Txnip and NLRP3, deactivates the NLRP3 inflammasome, and decreases myocardial infarct size.7 In a similar way, intramyocardial NLRP3 siRNA and inflammasome inhibitor both attenuate inflammatory cell infiltration and cardiomyocyte apoptosis in hearts that have been subjected to ischemia/reperfusion injury.7 In addition to the heart, the brain also exhibits Txnip-mediated inflammatory response under ischemic stress. In the brain, umbelliferone, a coumarin derivative with antioxidant properties, reduces Txnip expression and mitigates cerebral ischemia/reperfusion injury by suppressing NLRP3 inflammasome activation.8 Collectively, these data suggest a crucial role of inflammasomes in the pathophysiology of ischemia/reperfusion injury in which Txnip acts as a molecular switch that links redox balance with inflammatory response.
Thioredoxin-Interacting Protein Deficiency Improves Myocardial Glucose Utilization and Leads to Ischemic Tolerance
Another critical cellular function of Txnip is to serve as a glucostat. 18,20,24,63 Extracellular glucose stimulates a robust increase in the level of Txnip protein through a transcriptional mechanism. The induction of Txnip expression by glucose is mediated by a synergy between the tandem CCAAT box and carbohydrate response element motifs in the promoter region of the Txnip gene. 21,22,64 Thioredoxin-interacting protein, in turn, suppresses glucose uptake into the cell by binding directly to GLUT1, 24,25 which is a major mediator of basal glucose uptake in cardiomyocytes. The Txnip knockout cells have a higher capacity of glucose utilization through increased GLUT1 catalytic efficiency. 25 We previously generated cardiomyocyte-specific Txnip knockout mice 42 and directly measured glucose uptake in these animals. In cardiomyocyte-specific Txnip knockout mice hearts, we found a robust increase in myocardial glucose uptake compared to that of the littermate controls.
Energy utilization profiles are different between normal and ischemic hearts. Under normal physiological condition, the heart primarily utilizes free fatty acids with modest amounts of glucose in the fasting state at rest. However, in response to energetic stress such as myocardial ischemia, myocardial glucose utilization increases while fatty acid oxidation rate decreases. 65 The heart responds to oxygen deprivation by increasing glucose uptake and glycolysis. 66 Because lipids cannot be used as precursors for anaerobic metabolism, enhanced utilization of glucose becomes the primary means of supporting anaerobic metabolism under hypoxic conditions. The GLUT1 mRNA expression, which is constitutively expressed in cardiomyocyte sarcolemma, is increased up to two-fold in the ischemic regions of the myocardium in humans with coronary artery disease. 67 Although the overexpression of GLUT1 in mice results in unaltered contractile function at baseline, cardiac function exhibits better recovery in GLUT1-transgenic mice than in wild-type littermate control mice when their hearts are subjected to ischemia/reperfusion. 68 Thus, increased glucose uptake improves myocardial energetics, rendering the heart more resistant to ischemia/reperfusion injury. 68 Given the critical role of glucose supply in cardiac response to ischemia, it is plausible that the resulting increase in glucose delivery due to Txnip deficiency provides cardioprotection to the ischemic heart. Indeed, cardiomyocyte-specific deletion of Txnip in mice has been found to afford a protective advantage to the ischemic heart.1 Anaerobic metabolism with concomitant increase in glucose utilization following the deletion of Txnip preserves myocardial mechanical structure and function, thus promoting recoverability of the hypoxic heart. 1,42
Several pathways in molecular signaling of glucose metabolism regulated by Txnip have been proposed. The adenosine monophosphate-activated protein kinase (AMPK) is a metabolic sensor that monitors energy status to maintain cellular energy homeostasis. 69 The AMPK acts as a metabolic switch that is activated in response to a low energy status due to a depleted cellular ATP supply that occurs as a result of low glucose, hypoxia, and ischemia. The AMPK activation regulates signaling pathways that replenish cellular ATP supply and promote glucose uptake. 70 Recently, it has been reported that activation of AMPK causes phosphorylation of Txnip protein on Ser308, leading to its accelerated degradation.24 Under conditions of energetic stress, AMPK is activated and degrades Txnip protein, which in turn blocks Txnip-dependent inhibition of GLUT1, thus enhancing glucose uptake (Figure 3). Intrinsic activation of AMPK mediates ischemic glucose uptake and prevents postischemic cardiac dysfunction, apoptosis, and injury. 71,72 Thus, it is notable that AMKP works in coordination with Txnip to allow utilization of glucose as a fuel source to combat energy crisis due to ischemia/reperfusion injury in cardiomyocytes.

A hypothetical mechanism of cardioprotection by suppressing thioredoxin-interacting protein (Txnip) during ischemia. Ischemia causes energy stress. The adenosine monophosphate-dependent protein kinase (AMPK) is a sensor of energy status that is activated in response to cellular energy crisis. The AMPK promotes the phosphorylation and rapid degradation of Txnip protein, which leads to the suppression of Txnip. Glucose transporter 1 (GLUT1) facilitates the transport of glucose into the cell and is responsible for basal glucose uptake required to sustain energy homeostasis. Thioredoxin-interacting protein binds to GLUT1 and inhibits cellular glucose uptake. Conversely, Txnip suppression results in increased GLUT1 function, which promotes glucose uptake during ischemia. Enhanced glucose uptake is considered to support anaerobic metabolism and confer ischemic tolerance.
In addition to mediating glucose metabolism in response to ischemic stress, Txnip is also implicated in the autophagy pathway involved in ischemia/reperfusion injury. Conditions of stress, such as hypoxia, induce ROS through an upregulation of a protein complex involving regulated in development and DNA damage responses 1 (REDD1), an mTORC1 inhibitor. Thioredoxin-interacting protein forms a robust complex with REDD1 in response to energy crisis. The REDD1/Txnip complex mediates a stress-induced oxidative burst, which promotes autophagosome maturation that is crucial in the autophagy process. 73 Since autophagy plays a significant role in myocardial ischemia/reperfusion injury, 74 the interaction between Txnip and REDD1 may be involved in Txnip-mediated metabolic regulation in ischemia/reperfusion injury in the heart. Further studies are needed to determine the precise roles of these possible mechanisms.
Thioredoxin-Interacting Protein–microRNA Pathway May Control the Pathological Process of Ischemia/Reperfusion Injury
MicroRNAs (miRNAs) have emerged as key regulators of metabolic homeostasis. Ample data now suggest that miRNAs play a role in the pathophysiological condition of myocardial ischemia/reperfusion injury. 75 The miRNAs, a type of small noncoding RNA molecule containing 22 nucleotides, have implicated roles in negatively regulating gene expression in various physiological processes by binding to the 3′ untranslated region (3′UTR) of target genes. 76 -78 Recent data support a notion that miRNAs contribute to ischemia/reperfusion injury by modulating several key signaling pathways in cell division and cell survival. 79 Among the well-studied miRNAs, miR-135a, miR-34a, miR-155, miR-153, and miR-143 have shown to be regulators of cardiac repair in myocardial injury. 80 -82
Evidence from recent studies suggests that the role of Txnip in ischemia/reperfusion injury may involve an mRNAs-dependent pathway (Figure 4). Thioredoxin-interacting protein is found to be a potential target molecule of miR-135a since the overexpression of miR-135a protects HL-1 cells, a cardiac muscle cell line, from hypoxia/reoxygenation injury by downregulating Txnip. 83 Thioredoxin-interacting protein also controls myocardial fatty acid metabolism through an miRNA-mediated signaling pathway. Thioredoxin-interacting protein deficiency downregulates miR-33a, which results in an increased translation of critical enzymes for β-oxidation, thus supporting an elevated level of myocardial fatty acid oxidation. 84
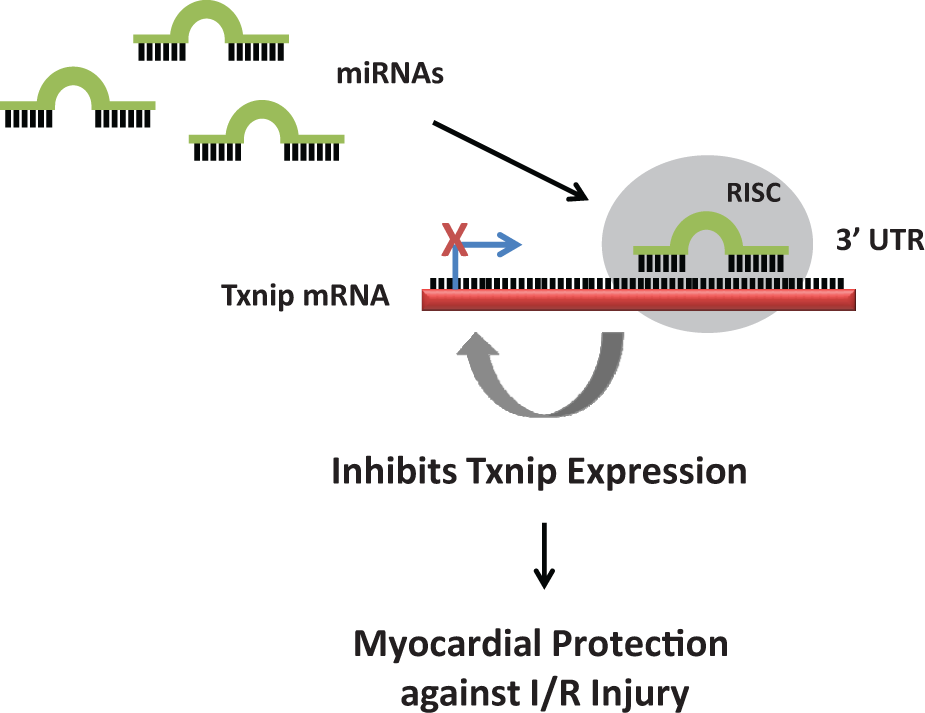
Small molecules called microRNAs (miRNAs) have been shown to influence the translation of thioredoxin-interacting protein (Txnip). The 3′ untranslated region (3′UTR) is the section of messenger RNA (mRNA) that follows the translation termination codon. The mature miRNA is incorporated into the RNA-induced silencing complex (RISC), the effector complex that targets the 3′UTR of Txnip mRNA. Binding of miRNAs to the 3′UTR of Txnip through base pairing either inhibits the expression or accelerates the degradation of Txnip mRNA, which in turn reduces the level of Txnip protein. Inhibition of Txnip expression can provide myocardial protection against ischemia/reperfusion (I/R), thus implicating the potential of miRNA-based therapy for ischemic heart disease.
Importantly, Txnip-miRNA pathway has now been suggested to play roles in diverse pathological conditions including diabetes and cancer. In the context of diabetes, Txnip induces miR-204 by inhibiting the activity of a signal transducer and activator of transcription 3 (STAT3), which then blocks insulin production in β cells. 85,86 Thioredoxin-interacting protein also downregulates miR-124a expression, which induces β-cell cytotoxicity. 86 In the context of cancer, miR-224/miR-452-mediated downregulation of Txnip is essential for E2F1-induced epithelial–mesenchymal transition (EMT) process and invasion of melanoma cells. 87 The expression of Txnip is also negatively correlated with the level of miR-373, which promotes the invasion and migration of breast cancer. 88 The MiR-373 suppresses Txnip by binding to the 3′UTR of Txnip, which in turn induces cancer cell EMT and metastasis. 89 In fact, activation of the miR-373-Txnip signaling pathway is associated with worse clinical outcomes in patients with breast cancer. 89 In addition to its clinical significance, the signaling pathway involving Txnip and miR373 is of particular interest also because miR-373 mediates glucose-induced cardiomyocyte growth, 90 which suggests the possibility that this pathway may contribute to cardiomyocyte survival in ischemia/reperfusion injury.
Furthermore, Txnip mRNA 3′UTR is predicted to be targeted by miR-20a. In a rat model of arthritis, miR-20a decreases the formation of NLRP3 inflammasome, suppresses the secretion of IL-1β, and promotes downregulation of Txnip in fibroblast-like synoviocytes. 91 Interestingly, it has been shown that overexpression of miR-20a inhibits hypoxia-mediated apoptosis, whereas knockdown of miR-20a induces cardiomyocyte apoptosis. 92 Thus, another possibility is that miR-20a may promote cardiomyocyte survival under myocardial ischemia/reperfusion by downregulating Txnip and deactivating NLRP3 inflammasome. However, these possibilities need to be validated in order to elucidate the exact roles of the Txnip-mRNA pathway in myocardial ischemia/reperfusion injury.
Conclusion
Although Txnip was initially characterized as a regulator of redox signaling through its interaction with thioredoxin, it is now clear that the functions of Txnip go beyond classical redox biology. Thioredoxin-interacting protein has been established as a key link among redox balance, inflammatory responses, and metabolic pathways. Thioredoxin-interacting protein is also involved in other signaling pathways that are not covered in this review but summarized elsewhere. 16,93,94 Clearly, some progress has been made towards understanding of the mechanisms that regulate Txnip during ischemia and energetic stress. First, Txnip is a negative regulator of thioredoxin and its reducing capacities. In ischemia/reperfusion injury, Txnip is associated with harmful consequences by promoting oxidative damage in tissues via its pro-oxidative effects. Second, the activation of NLRP3 inflammasome by Txnip can be important in the development of tissue damage induced by ischemia/reperfusion. Third, under ischemia/reperfusion injury, AMPK may be activated to degrade Txnip protein, which in turn blocks Txnip-dependent inhibition of GLUT1, thus allowing enhanced glucose uptake to relieve energy crisis. Fourth, the role of Txnip in ischemia/reperfusion injury may involve an mRNA-dependent pathway. Although these findings highlight the critical connection between Txnip and ischemic stress, the complexity of Txnip’s role in ischemia/reperfusion injury warrants further investigation.
Even though compelling human evidence is currently lacking, Txnip suppression is generally associated with a protective advantage against ischemia/reperfusion injury. A number of currently available therapeutic agents for human diseases have shown to have the potential to control Txnip expression to some degree. These agents include calcium channel blockers, 95,96 angiotensin-converting enzyme inhibitors, 6,97 angiotensin-II receptor blockers, 98 glucagon-like peptide-1 agonists, 99 and insulin. 20 To date, however, there is no pharmacological therapy specifically designed to inhibit Txnip expression. Thus, the interaction between Txnip and miRNAs is of great interest for the development of novel therapeutic entities. Additional studies using a combination of molecular and physiological approaches will provide an integrated understanding for future targeted therapies that harness pharmacological or genetic control of Txnip.
Footnotes
Authors’ Contribution
Bing F. Wang contributed to conception and design, contributed to acquisition, analysis, and interpretation, critically revised the manuscript, and agrees to be accountable for all aspects of work ensuring integrity and accuracy. Jun Yoshioka contributed to conception and design, contributed to acquisition, analysis, and interpretation, drafted the manuscript, gave final approval, and agrees to be accountable for all aspects of work ensuring integrity and accuracy.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by awards from American Heart Association (13GRNT16870007), Philip V. and Anna S. Brown Foundation, Bank of America, N.A., Trustee, National Institute of Health (1R01 HL130861) and Watkins Discovery Award.