
Abstract
The present study further identified factors involved in the cardioprotective phenomenon of remote preconditioning of trauma (RPCT) with special emphasis on the role of the epoxyeicosatrienoic acids (EETs) in mediating this phenomenon. Remote preconditioning of trauma was produced by an abdominal incision only through the skin. Subsequently, all rats were subjected to 30 minutes of left coronary artery occlusion followed by 2 hours of reperfusion and the infarct size was determined. Remote preconditioning of trauma produced a reduction in infarct size expressed as a percentage of the area at risk from 63.0% ± 1.1% to 44.7% ± 1.4%; P < .01 versus control. To test the 3 major triggers of classical preconditioning in mediating RPCT, blockers of the bradykinin B2 receptor (B2BK), (S)-4-[2-[Bis(cyclohexylamino)methyleneamino]-3-(2-naphthalenyl)-1-oxopropylamino]benzyl tributyl phosphonium (WIN 64338, 1 mg/kg, iv), or HOE 140 (50 μg/kg, iv), the nonselective opioid receptor blocker, naloxone (3 mg/kg, iv), or the adenosine A1 receptor blocker, 8-Cyclopentyl-1,3-dipropylxanthine (DPCPX, 1 mg/kg, iv) were administered 10 minutes prior to RPCT. Only the 2 B2BK selective antagonists blocked RPCT (60.2% ± 1.1%, WIN 64338; 62.3% ± 2.0%, HOE 140). To test EETs in RPCT, we administered the EET receptor antagonist 14,15-Epoxyeicosa-5(Z)-enoic acid (14,15-EEZE, 2.5 mg/kg, iv) or the EET synthesis inhibitor, N-(Methylsulfonyl)-2-(2-propynyloxy)-benzenehexanamide (MSPPOH, 3.0 mg/kg, iv) 10 minutes prior to RPCT. In both groups, the EET antagonists completely blocked RPCT (62.0% ± 0.8%, 14,15-EEZE; 61.8% ± 1.0%, MSPPOH). The EET antagonists also blocked the effect of B2BK activation. We also determined whether the sarcolemmal KATP or the mitochondrial KATP channel mediate RPCT by pretreating rats with 1-[5-[2-(5-Chloro-o-anisamido)ethyl]-2-methoxyphenyl]sulfonyl-3 methylthiourea, sodium salt (HMR 1098) or 5-hydroxydecanoic acid (5-HD), respectively. Interestingly, 5-HD blocked RPCT (64.7% ± 1.3%), whereas, HMR 1098 did not (50.3% ± 1.3%). The 2 EET antagonists completely blocked capsaicin-induced cardioprotection. These results clearly suggest that EETs mediate RPCT-, bradykinin- and capsaicin-induced cardioprotection in rat hearts.
Introduction
Previous work from several laboratories, including ours, demonstrated that remote preconditioning of trauma (RPCT) produced by a nonischemic abdominal wall surgical incision markedly reduced infarct size (IS) in mice and dog hearts. 1,2 Evidence suggests a sensory neural pathway is involved with spinal nerves and sympathetic nerves being activated. Bradykinin, PKC∊, the mitochondrial (mito) KATP channel, and the epoxyeicosatrienoic acids (EETs, the cytochrome P450 epoxygenase metabolites of arachidonic acid) were also triggers or mediators of RPCT in mice (BK, PKC, mito KATP) and dogs (EETs) and topical capsaicin mimics RPCT. These results strongly suggest that RPCT is neuronally mediated; however, one cannot rule out circulating humoral factors such as adenosine and opioids being involved 3 as shown in other models of remote ischemic preconditioning (RIPC) produced by activation of mesenteric nerves and skeletal muscle. 4,5 Furthermore, the role of the EETs in RPCT and species other than the dog is not clear. This RPCT phenomenon has not been previously characterized in the rat. Therefore, the present study was designed to determine in a rat model of infarction the major players mediating RPCT and more clearly defined the role of EETs and KATP channel subtypes in mediating cardioprotection in this model.
Materials and Methods
Materials
Both 14,15-Epoxyeicosa-5(Z)-enoci acid (14,15-EEZE) and N-(Methylsulfonyl)-2-(2-propylnyloxy)-benzenehexanamide (MSPPOH) were synthesized in the laboratory of J.R.F. 5-Hydroxydecanoic acid (5-HD), naloxone, HOE 140, glibenclamide, bradykinin, and 8-Cyclopentyl-1,3-dipropylxanthine (DPCPX) were obtained from Sigma Chemical Company (St Louis, Missouri). (S)-4-[2-[Bis(cyclohexylamnio)methyleneamino]-3-(2-naphthalenyl)-1-oxopropylamino]benzyl tributyl phosphonium (WIN 64338) were obtained from Tocris Bio-Sciences (Ellesville, Missouri). 1-[5-[2-(5-Chloro-o-anisamido)- ethyl]-2-methoxyphenyl]sulfonyl-3-methylthiourea, sodium salt (HMR 1098) was kindly provided by Dr Hans Gogelein, Aventis Pharmaceuticals, Germany. Capsaicin cream was obtained over the counter. Both 14,15-EEZE and MSPPOH were dissolved in a vehicle composed of 95% ethanol, polyethylene glycol 200, and 1 N sodium hydroxide (5:5:1). Naloxone, HOE 140, 5-HD, HMR 1098, and bradykinin were dissolved in distilled water. WIN 64338, DPCPX, and glibenclamide, were dissolved in DMSO (0.01%).
General Preparation of Rats
All experiments were conducted in accordance with the “Position of the American Heart Association on Research and Animal Use” adopted by the American Heart Association and the guidelines of the Biomedical Resource Center and the IACUC Committee of the Medical College of Wisconsin. The Medical College of Wisconsin is accredited by the American Association of Laboratory Animal Care (AALAC).
The protocol for the rat preparation and experiments has been previously described in detail. 6 Male Sprague-Dawley rats (250-300 g) were anesthetized with 100 mg/kg of Inactin, placed on a heating pad and a tracheotomy performed and the rat ventilated with room air supplemented with 100% oxygen. Catheters were placed in the carotid artery and jugular vein for measurement of systemic blood pressure, heart rate, blood gases, and for administration of drugs or vehicle. The heart was exposed by a left thoracotomy and the left coronary artery isolated and a suture placed around it to produce an occlusion followed by reperfusion. The hearts were subjected to 30 minutes of total coronary artery occlusion followed by 2 hours of reperfusion at which time the artery was reoccluded and Evans Blue Dye injected into the jugular vein to stain the nonischemic area of the heart blue. Subsequently, the heart was arrested with a bolus of 10% KCl and removed for the measurement of IS. After removing the heart, it was washed in buffer and cut in a bread loaf fashion into 5 to 6 pieces and the blue-stained normal region and the ischemic region were separated and placed in separate vials containing triphenyltetrazolium chloride staining solution and incubated at 37°C in phosphate buffer at pH 7.40 for approximately 15 minutes. Subsequently, the pieces of tissue were placed into vials containing formaldehyde and stored overnight at room temperature and IS determined by gravimetry the following day. Infarct size was expressed as a percentage (%) of the area at risk (IS/AAR, %) as determined by our dual staining technique. In the AAR, the dark red color represents noninfarcted tissue, whereas the infarcted tissue remains pale gray.
Experimental Design
Rats were randomly assigned to 30 groups for the different treatments and 8 rats were usually assigned to each group of experiments. In all studies, rats were subjected to 30 minutes of left coronary artery occlusion (LCAO) and 2 hours of reperfusion. In all slit experiments, the abdominal slit was performed 15 minutes prior to the 30-minute occlusion period. A 4-cm transverse skin incision was made via the abdominal midline of the rats with a scalpel similar to the previously described protocols. 1,2 The pharmacological antagonists or appropriate vehicle were administered alone 15 minutes preocclusion in control experiments and in the slit experiments (RPCT) the various antagonists were administered intravenously (iv) 15 minutes prior to RPCT (Figure 1). In some experiments, bradykinin was administered iv 15 minutes prior to the 30-min occlusion period and the respective receptor antagonist, WIN 64338 or in some experiments the EET antagonists were administered 15 minutes prior to bradykinin. Finally, the same amount of external capsaicin cream (0.1%) was administered topically on the same surface area at the site of the slit. In 3 groups, the EET antagonists, 14,15-EEZE and MSPPOH, were given iv 15 minutes prior to capsaicin cream application.

Experimental protocol for remote preconditioning with trauma (RPCT) in a rat in vivo infarct model.
Statistical Analysis
All values are expressed as the mean ± standard error of the mean (SEM). Differences between groups in hemodynamics and blood gases were compared by the use of a 2-way analysis of variance (ANOVA) followed by Tukey post hoc test. Differences between groups in AAR, IS, and IS/AAR were compared by a 1-way ANOVA. Differences between groups were considered significant if P < .05.
Results
Hemodynamics and Blood Gases
Heart rate, systolic and diastolic blood pressure, and blood gases (data not shown for blood gases) at baseline, at 30 minutes of occlusion or at 2 hours of reperfusion were not significantly different between groups (Tables 1 and 2). There were also no differences in the AAR as a percentage of the left ventricle ([LV] AAR/LV; Table 1). These data suggest that differences in IS were not different as a result of changes in hemodynamics or AAR.
Hemodynamic Valuesa,b

Abbreviations: AAR, area at risk; Con, control; LV, left ventricle; Occ, occlusion; Rep, reperfusion; SEM, standard error of the mean; RPCT, remote preconditioning with trauma; 5-HD, 5-hydroxydecanoic acid.
aHeart rate and AAR/LV.
bAll values are the mean ± SEM (N = 8-15 rats).
Hemodynamic Valuesa,b

Abbreviations: Con, control; Occ, occlusion; Rep, reperfusion; DBP, diastolic blood pressure; SBP, systolic blood pressure; RPCT, remote preconditioning with trauma; 5-HD, 5-hydroxydecanoic acid; SEM, standard error of the mean.
aBlood pressure, mm Hg.
bAll values are the mean ± SEM (N = 8-15 rats).
Mediators of Classical Ischemic Preconditioning
To determine the role of the 3 G protein-coupled receptors that have been demonstrated to trigger classical ischemic preconditioning (IPC), adenosine, 7 opioids or bradykinin B2 receptors, 8 specific antagonists of these 3 receptors were administered alone 15 minutes prior to the 30-minute occlusion period (no slit control) or 15 minutes prior to RPCT and IS/AAR are determined (Figure 2). Initially, we showed that the transverse abdominal slit resulted in a significant reduction in IS/AAR from 63.0% ± 1.1% to 44.7% ± 1.4% (P < .01). The reduction of IS/AAR produced by RPCT is much less than the reduction we have previously published for IPC (IS/AAR = 11.0% ± 3.0%). 6 Administration of DPCPX, the adenosine A1 receptor antagonist, naloxone, the nonselective opioid receptor antagonist or WIN 64338 or HOE 140, 2 bradykinin B2 receptor antagonists alone in the absence of the abdominal slit had no effect on IS/AAR. However, the 2 B2 receptor antagonists, WIN 64338 or HOE 140, blocked the protective effect of RPCT (63.2% ± 1.5%, 61.7% ± 1.3%), whereas DPCPX (45.4% ± 2.2%) and naloxone (44.5% ± 1.2%) had no effect on RPCT-induced protection (Figure 2). These results indicate that bradykinin/B2 receptor pathway is a major signaling in the RPCT-induced cardioprotection. The results also suggest the signaling pathway for RPCT is different from classical or remote preconditioning produced by a brief ischemic insult.

Remote preconditioning with trauma (RPCT) results in a significant reduction in myocardial infarct size (IS) expressed as a percentage of the area at risk (IS/AAR) in the intact rat heart. This effect was abolished by pretreatment with the selective bradykinin 2 receptor (BK2R) antagonists, WIN 64338 (1.0 mg/kg, intravenously [iv]) and HOE 140 (50 μg/kg, iv) but not by the adenosine A1 receptor blocker, DPCPX (1.0 mg/kg, iv) or by the nonselective opioid receptor antagonist naloxone (3.0 mg/kg, iv) All values are the mean ± standard error of the mean (SEM) of 8 animals/group. *P < .01 vs the control group.
We previously 2 showed that bradykinin was a mediator of RPCT in the dog heart since RPCT was blocked by HOE 140 and the EETs were also important players in RPCT since the EET receptor antagonist, 14,15-EEZE, and the EET synthesis inhibitor, MSPPOH, both blocked RPCT in the canine heart. Therefore, we tested the hypothesis that the EETs were downstream mediators of the B2 receptor. Administration of bradykinin (60 μg/kg, iv) 15 minutes prior to the 30-minute ischemic period produced a significant reduction in IS/AAR (46.9% ± 1.2%; P < .01) similar in magnitude to that seen with RPCT (Figure 2). This protective effect of bradykinin was completely abolished by 14,15-EEZE (62.9% ± 1.1%) and MSPPOH (61.6% ± 1.8%) which suggests that bradykinin released by RPCT is exerting its cardioprotective effect via the EETs in rat hearts (Figure 3) similar to that previously shown in the dog heart. 2
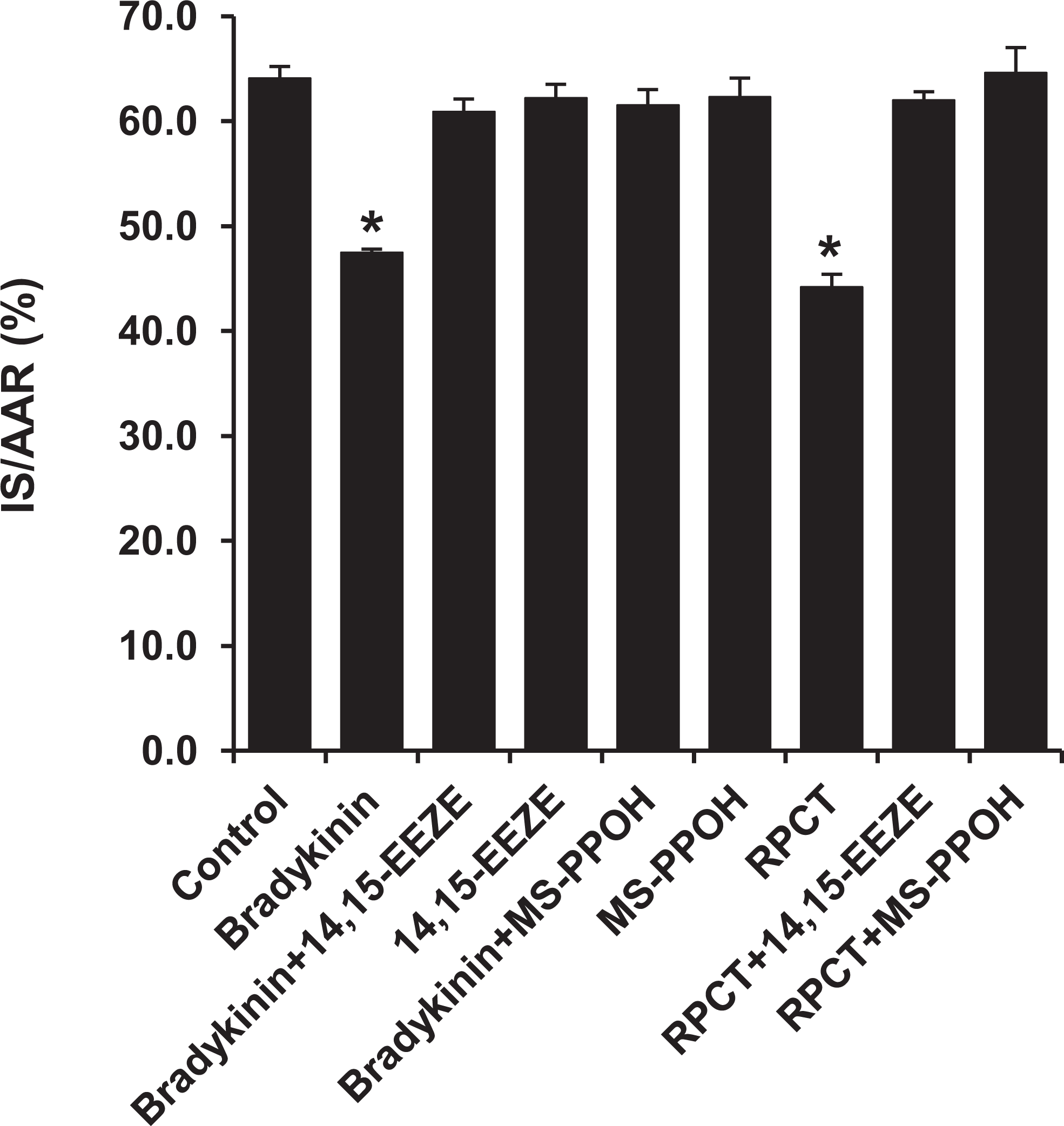
Bradykinin (60 μg/kg, iv) produced a significant reduction in IS/AAR in the intact rat heart. This cardioprotective effect was abolished by pretreatment with the EET receptor blocker, 14,15-EEZE (2.5 mg/kg, iv), and the EET synthesis inhibitor, MSPPOH (3.0 mg/kg, iv). These effects of bradykinin and 14,15-EEZE and MSPPOH in cardioprotection are identical to the RPCT-mediated cardioprotection. All values are the mean ± SEM of 8 animals/group. *P < .01 vs the Control Group. EET indicates epoxyeicosatrienoic acid; IS, infarct size; AAR, area at risk; iv, intravenously; RPCT, remote preconditioning with trauma; SEM, standard error of the mean.
Role of EETs in Capsaicin-Induced Cardioprotection and RPCT
To further determine a role for sensory C-fibers in mediating RPCT in rat hearts, capsaicin cream (0.1%) was applied at the same position as the abdominal slit 15 minutes prior to the 30-minute ischemic period. In agreement with previous results obtained in the mouse heart, 1 capsaicin produced a significant reduction in IS/AAR (47.5% ± 0.3%; P < .01). The effect of capsaicin cream was completely abolished by pretreatment with either of the 2 EET antagonists, 14,15-EEZE (60.9% ± 1.2%) or MSPPOH (61.5% ± 1.5%; Figure 4).

Topical application of capsaicin cream (0.1%) produced a significant reduction in IS/AAR similar in magnitude to that observed with RPCT (Figure 2). The cardioprotective effect of capsaicin cream was completely abolished by pretreatment with the 2 EET antagonists, 14,15-EEZE (2.5 mg/kg, iv) or MSPPOH (3.0 mg/kg, iv). All values are the mean ± SEM of 8 rats/group. *P < .01 vs the control group. EET indicates epoxyeicosatrienoic acid; IS, infarct size; iv, intravenously; AAR, area at risk; RPCT, remote preconditioning with trauma; SEM, standard error of the mean.
Role of Sarcolemmal or Mitochondrial KATP Channel in RPCT
Previous data published by Jones et al 1 showed that the cardiac KATP channel is a distal mediator of RPCT in the mouse heart, however, the subtype involved, sarc KATP channel or the mito KATP channel or both was not addressed. Therefore, we determined whether the sarc KATP or the mito KATP channel was involved in RPCT by pretreating rats with glibenclamide, a nonselective KATP channel antagonist, HMR 1098, a selective sarc KATP antagonist, or 5-HD, a selective mito KATP blocker. Interestingly, glibenclamide and 5-HD completely blocked the protective effect of RPCT (61.7% ± 1.3%, 64.7% ± 1.3%), whereas, HMR 1098 did not (50.3% ± 1.3%; Figure 5). None of the 3 KATP channel antagonists had any effect on IS/AAR when administered to rats without the abdominal slit (Figure 5).
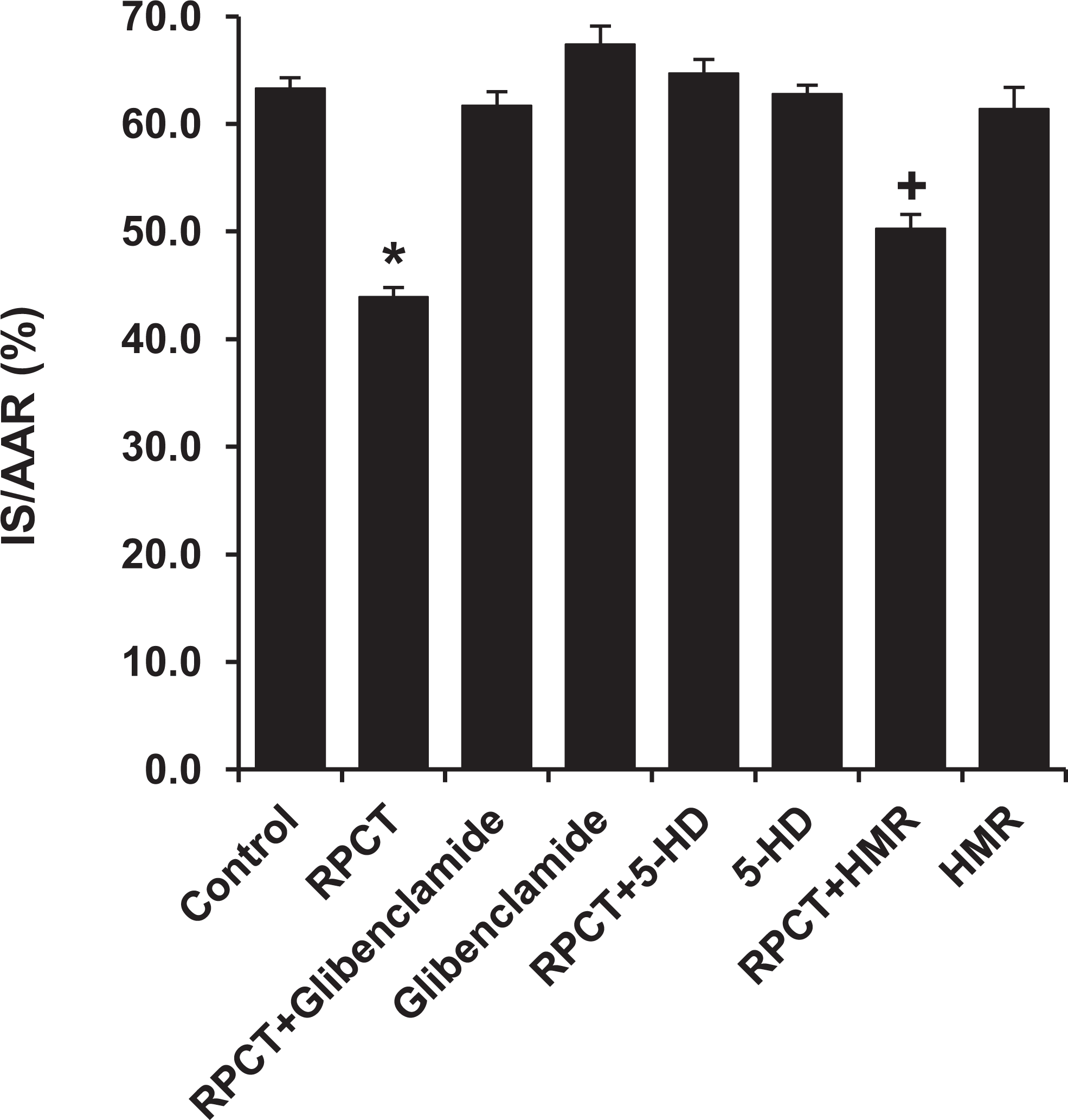
Pretreatment with the nonselective KATP channel antagonist, glibenclamide (1.0 mg/kg, iv), and the selective mito KATP channel antagonist 5-HD (10 mg/kg, iv) completely abolished the cardioprotective effect of RPCT, whereas the selective sarcolemmal KATP channel blocker, HMR 1098 (6.0 mg/kg, iv), did not block the beneficial effect of RPCT. All values are the mean ± SEM of 8 rats/group. *P < .01 vs the control group. + P < .05 vs the control group. RPCT indicates remote preconditioning with trauma; SEM, standard error of the mean.
Discussion
The results of the present study are the first to characterize the signaling pathways responsible for RPCT in the rat heart. The data suggest that, similar to results obtained for the mouse 1 and dog 2 heart, the sensory nerve activation produced by a surgically induced abdominal incision or by topically applied capsaicin results in a marked reduction in myocardial IS. The cardioprotective effects of the nonischemic surgical trauma by abdominal incision have been attributed to the activation of skin nociceptors and require neurogenic signaling and activation of cardiac sensory and sympathetic nerves. 1 It is unclear whether thoracotomy per se contributes to cardioprotection. Nonetheless, the effect of thoracotomy (if any) is included in the IS of the control rats. The reduction in IS in rats with abdominal incision as compared with the control rats was obtained from groups of rats with the same thoracotomy. In the nonischemic surgical groups produced by abdominal incision, bradykinin appears to be the common mediator of cardioprotection via activation of the B2 bradykinin receptor. In all 3 animal species, bradykinin receptor antagonists, HOE 140 or WIN 64338, blocked the protective effect of RPCT and also blocked the cardioprotective effect of exogenously administered bradykinin.
Since bradykinin, adenosine, and endogenous opioids are the 3 major G protein-coupled receptors 7,8 involved in mediating classical IPC and several models of ischemically induced RPC, 4,5,9 we examined a role of these 3 classical mediators in mediating RPCT by using selective receptor antagonists. Interestingly, bradykinin was the only mediator involved in RPCT in the rat heart, making this pathway different from classical IPC and RPC. To further confirm a role of bradykinin, exogenously administered bradykinin produced a reduction in IS similar in magnitude to that produced by RPCT (Figure 3).
Previous work from our laboratory 10,11 in the canine heart demonstrated an important role for the EETs as mediators of both IPC and postconditioning. Furthermore, the cardioprotective effect of bradykinin 2 in dog hearts was abolished by 2 EET antagonists, 14,15-EEZE, 12 a putative EET receptor antagonist, and MSPPOH, an EET synthesis inhibitor. 13 Similar experiments were performed in the rat heart with exogenous bradykinin and the 2 EET antagonists with confirming results. Bradykinin mimicked RPCT-induced IS reduction blocked by 14,15-EEZE and MSPOH (see Figure 3). These findings suggest an important role for EETs in mediating RPCT as part of the bradykinin signaling pathway.
Another stimulus mimicking the effect of RPCT in the mouse heart is the sensory nerve activator, capsaicin, which was shown by Jones et al 1 to mimic RPCT when applied topically to the same area as used for the abdominal slit stimulation. On topical application of 0.1% capsaicin cream to the abdomen, we obtained a reduction in IS similar to that produced by RPCT. The cardioprotective effect of capsaicin cream was completely abolished by 14,15-EEZE or MSPPOH which further supports a key role for the EETs in mediating RPCT. More experiments are needed to determine the source of the EETs in mediating RPCT and where these compounds are spatially located in the signaling pathway.
In a final series of experiments, we determined the subtype of KATP channel, which mediates RPCT. In the initial study published by Jones et al, 1 RPCT in mice was partially blocked by the nonselective KATP channel antagonist, glibenclamide, and was completely abolished by the putative mito KATP channel antagonist 5-HD. In the rat heart, glibenclamide and 5-HD both completely abolished the cardioprotective effect of RPCT, whereas the selective sarcolemmal (sarc) KATP antagonist did not block RPCT (Figure 5). Although it is surprising that glibenclamide did not abolish RPCT in the mouse, whereas 5-HD did, 1 our results clearly support the mito KATP channel as the major mediator of RPCT in rats. In spite of these results obtained with HMR 1098 and 5-HD, caution should be used when interpreting data obtained using these 2 KATP channel antagonists. There is considerable controversy surrounding the use of HMR 1098 and 5-HD as selective blockers of the sarc and mito KATP channels, and several published papers suggest the KATP blockers possess alternative modes of action. 14,15 Nevertheless, the data obtained with glibenclamide, a well-known sulfonylurea KATP blocker, 16 strongly suggest a role for cardiac KATP channel in mediating RPCT in the rat and mouse heart, most likely, the mito KATP channel.
We were surprised that naloxone did not block RPCT in the current study since we previously showed that opioids mediate the cardioprotective effect of exogenously administered EETs in rat hearts. In both studies, naloxone itself had no effects on IS of the rat heart. However, naloxone abolished the reduction in infract size mediated by exogenous 11,12- or 14,15-EET in the intact rat heart 17 but did not block the reduction in IS produced by RPCT (present study). At the moment, we have no easy answer for these puzzling observations. We speculate that the RPCT may release multiple classes of mediators, particularly bradykinin as supported by our current results (Figure 2). Bradykinin stimulates the production of EETs in a variety of systems, 18,19 and this hypothesis is supported by our results that the effects of both RPCT and bradykinin in cardioprotection are blocked by 14,15-EEZE and MSPPOH (Figures 2 and 3). The EETs can activate numerous downstream signaling pathways including activation of endothelial nitric oxide synthase to release nitric oxide 20,21 and vascular and cardiac KATP channels. 22,23 Our current data suggest that at least the mito-KATP channel is involved in the RPCT-mediated cardioprotection (Figure 5). The RPCT model possesses several questions of where the release of EETs occurs and whether the RPCT influences the downstream signaling of the EETs in which opioids may not be a major participant. Thus, the current results from RPCT may be different from those obtained following the direct administration of the EETs. Measurements of EETs, opioids, and bradykinin in hearts or plasma may help furnish the answer.
Unanswered Questions and Future Directions
There are several questions which remain in characterizing the phenomenon of RPCT. Obviously, this is a real endogenous cardioprotective mechanism which has been confirmed to exist in 3 animal species thus far, mouse, rat, and dog with many similarities in the signaling components investigated. One area of controversy that exists concerns the magnitude of the IS reduction. The greatest reduction in IS/AAR occurs in mouse (approximately 80%), followed by dog (approximately 60%) and then rat (approximately 30%). It is not clear as to why there is such a large variability between these 3 species. Perhaps a better and more complete understanding of the neural and humoral 9 signaling pathways involved in these species will lead to a novel compound that will cause a reduction in IS equivalent to that observed in mouse heart. Another interesting finding in the rat and dog studies is the important role played by the CYP epoxygenase pathway in mediating RPCT. Thus far, the evidence for an important role of the EETs in mediating RPCT is based on the use of 2 antagonists, 14,15-EEZE and MSPPOH. To confirm the importance of this pathway in mediating RPCT, it will be necessary to measure EET concentrations in plasma or cardiac tissue prior to and following RPCT, bradykinin, or capsaicin administration to demonstrate that these stimuli actually increase EET concentrations and that these EET blockers antagonize these effects. As always in research, there are more questions than answers. At present, the RPCT phenomenon may be most important since its signaling pathway provides valuable new insight into cardioprotective signaling that is worth studying, with the hope of an eventual lead to a clinical application. 3
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by funds obtained from NIH grant HL 74314 (GJG).