
Abstract
Estrogens have been recognized, in the last 3 decades, as important hormones in direct and indirect modulation of vascular health. In addition to their direct benefit on cardiovascular health, the presence of esterified estrogen in the lipid core of high-density lipoprotein (HDL) particles indirectly contributes to atheroprotection by significantly improving HDL quality and functionality. Estrogens modulate their physiological activity via genomic and nongenomic mechanisms. Genomic mechanisms are thought to be mediated directly by interaction of the hormone receptor complex with the hormone response elements that regulate gene expression. Nongenomic mechanisms are thought to occur via interaction of the estrogen with membrane-bound receptors, which rapidly activate intracellular signaling without binding of the hormone receptor complex to its hormone response elements. Estradiol in particular mediates early and late endothelial nitric oxide synthase (eNOS) activation via interaction with estrogen receptors through both nongenomic and genomic mechanisms. In the vascular system, the primary endogenous source of nitric oxide (NO) generation is eNOS. Nitric oxide primarily influences blood vessel relaxation, the heart rate, and myocyte contractility. The abnormalities in expression and/or functions of eNOS lead to the development of cardiovascular diseases, both in animals and in humans. Although considerable research efforts have been dedicated to understanding the mechanisms of action of estradiol in regulating cardiac eNOS, more research is needed to fully understand the details of such mechanisms. This review focuses on recent findings from animal and human studies on the regulation of eNOS and HDL quality by estradiol in cardiovascular pathology.
Keywords
Introduction
Female sex steroid hormones exert diverse effects on the vascular system, inducing rapid vasodilatation, reducing vessel wall's response to injury, and decreasing the development of atherosclerosis. 1 –3 These effects are the result of a multifaceted series of actions on the various components of the vascular wall, from endothelial cells (ECs) to vascular smooth muscle cells (VSMCs), to macrophages and stromal vascular cells, deriving by a complex interplay of estrogen receptor (ER)-mediated genomic as well as nongenomic mechanisms. 1 The vasculature is the site of critical changes with aging and estradiol loss, including increased atherosclerosis and abnormal function (relaxation and constriction). 4 Endothelial cells, as well as VSMCs, are important targets of estradiol, and changes in estradiol and endothelial nitric oxide synthase (eNOS) in particular have an impact on the vasculature. 4 The arterial vasodilation is rapidly induced by estradiol, primarily by activating nitric oxide synthase (NOS) in EC. 5 Prevention of dynamic constriction or spasm of coronary arteries is possible by enhanced vasodilation of arteries as a result of augmented release of nitric oxide (NO) by estradiol. 6
Nitric oxide plays a critical role in the regulation of cardiovascular function via endogenously produced or exogenously applied NO, such as NO donor. 7 Nitric oxide influences blood vessel relaxation, and heart rate in a way of modulating VSMC and myocyte contractility. 7,8 Nitric oxide-mediated blood vessel relaxation is achieved by activating soluble guanylyl cyclase (sGC) and increasing intracellular levels of cyclic guanosine monophosphate (cGMP). 9 Nitric oxide is a highly reactive signaling molecule and a critical modulator of myocardial function. 10 Three distinct genes are known to code for the different NOS isoforms, 10 namely, eNOS (NOS III), neuronal NOS (nNOS, NOS I), and inducible NOS (iNOS, NOS II). 11 Tissue and cellular distributions of these isoforms coupled with different levels of expression and cofactor requirements in each tissue modulate their specific functions in vivo. 10 In the heart, all 3 NOS isoforms are present: nNOS and eNOS are constitutively present in distinct subcellular locations within cardiomyocytes, whereas iNOS is absent in the healthy heart, but its expression is induced by proinflammatory mediators. 10 Endothelial NOS isoform is the primary endogenous source of NO generation in the vascular system. 12,13
Fundamental cardiovascular functions are regulated by the steroid hormones 14 , though there are marked gender differences. 14 Cardiomyocytes, VSMCs, and ECs are all well equipped with sex steroid hormone receptors as well as with the steroid metabolizing enzyme aromatase. 14 Human arteries and veins express aromatase. Aromatase converts testosterone into estradiol. 14 Aromatase deficiency accelerates atherogenesis, especially in men. 14 Modifications of cardiovascular function in women are associated with changes in estrogen concentrations. 14 It was observed that arteries from elderly women were less responsive than arteries from men, suggesting these sex differences persist after menopause. 15 Therefore, lower incidence of stroke in women compared to men is seen until the age of 85 years. 15,16 In comparison with men, women get their first stroke about 4.5 years later; however, morbidity rises after menopause and, with age, reaches that of men. 16,17 Mechanisms underlying male/female differences in human arteries may not require continued presence of sex hormones but instead result from the influences of sex chromosomes and/or organizational effects of sex hormones that commit tissues to a male or female phenotype. 15,18 Estradiol is an important sex steroid hormone in human and animal physiology with pleotropic functions. 19,20 These physiological processes include development, growth, metabolism, reproduction, and sexual function in both humans 19 –21 and animals. 19,20,22,23 In the heart and peripheral blood vessels, estradiol induced rapid arterial vasodilation, 20,24,25 inhibition of atherosclerotic lesions, 20,24,26 cardioprotective effect following trauma-hemorrhage, 20,27 and amelioration of ischemia/reperfusion-induced myocardial injury. 20,28
The beneficial vascular effects of estradiol have been ascribed to genomic and nongenomic effects on the blood vessels. 29 –31 Genomic effects of estradiol include stimulation of EC growth and inhibition of VSMC proliferation. 29,30,32 Estradiol causes rapid nongenomic vasodilation of blood vessels via activation of endothelium-dependent vascular relaxation pathways. 29,33,34 The positive protective effects of eNOS in cardiovascular system (CVS) are that it promotes vasodilatation of VSMC, inhibits smooth muscle cell proliferation, and decreases platelet adhesiveness. 35,36 Endothelial NOS also decreases adhesiveness of the endothelial layer to the white blood cells and acts as an anti-inflammatory agent. 35 Increased vasodilatation and diminished response of blood vessels to injury are implicated as estradiol effects. 3 In this review, we will focus on discussion of the potential role of estradiol in regulating eNOS activity and its function in the vascular system. Also, in this review, we will initially focus on the direct effects of estrogen on vascular health with emphasis on the regulation of eNOS activity in endothelial and VSMCs and then on the effects of estrogens on high-density lipoprotein (HDL) quality and functionality pertinent to atheroprotection. Understanding the atheroprotective functions of estrogens may contribute to the development of novel therapeutic strategies for the treatment of cardiovascular diseases (CVDs).
General Mechanisms of Estrogen Regulation of eNOS
Estrogens are the principal sex steroid hormones produced in the ovaries, placenta, breast, and peripheral adipose tissues in women as well as in testis and peripheral adipose tissues in men. 37,38 Plasma levels of estradiol in women are approximately 4 times higher than those detected in men. 38,39 In vivo produced estrogens encompass estradiol, estriol, and estrone; however, estradiol is considered the most potent estrogen hormone. 40,41
Estrogen hormones modulate cellular function via genomic 42,43 and nongenomic mechanisms. In the genomic signaling pathway, the estradiol binding to its receptors results in receptor activation and transformation, leading to receptor dimerization. Upon binding of the hormone, these receptors function as ligand-inducible transcription factors by binding to a specific DNA element known as estrogen response element (ERE). 38,44 This interaction results in recruitment of a host of activators or silencers and regulated gene transcription. 42 Genomic mechanism generally has a longer onset and duration of action (Figure 1A). 42,43 The magnitude of the temporal relationship of nuclear transcriptional regulation is dependent on the formation of the appropriate complex of ER with other coactivators and/or transcriptional factors. 42

Schematic presentation of: (A) genomic and (B) nongenomic effects of E2 on eNOS activity/expression. E2 indicates estradiol; ERα, estrogen receptor α; ERβ, estrogen receptor β; ERE, estrogen response elements; mRNA, messenger ribonucleic acid; eNOS, endothelial nitric oxide synthase; PI3K, phosphatidylinositol 3-kinase; Akt, protein kinase B; NO, nitric oxide; p-Akt/P-eNOS, phosphorylated form of Akt/eNOS; GPR30, G protein-coupled receptor 30; SR-BI, scavenger receptor B, type I.
In addition to the classical genomic mechanism, it has been documented that estrogens can modulate the cellular function via interaction with membrane receptors and regulate cellular signaling independent of direct binding to EREs (Figure 1B). 42,45 This mechanism is known as the rapid signaling pathway. 42 This pathway is attributed to the binding of estradiol to the membrane ER and the activation of phosphatidylinositol 3-kinase (PI3K)/protein kinase B (Akt) pathway resulting in eNOS phosphorylation, 46,47 and increased eNOS activity. The rapid signalling mechanism is also referred to as nongenomic pathway. The molecular mechanism of action of estrogens is believed to be mediated by the classical ER family (ERα and ERβ). 13,44,48 Estrogen receptors are expressed in a variety of tissues and cells. In the absence of the hormone, these receptors are inactive. Although the exact nature of the ER on the membrane remains controversial, membrane-associated ERα and/or ERβ mediating eNOS phosphorylation at Ser1177 via activation of the PI3K/Akt pathway, that leads to increased eNOS activity, has been reported. 13,45,47 Both genomic and nongenomic mechanisms are involved in the regulation of eNOS activity and function by estradiol in cardiovascular tissues. 42
In recent years, a third membrane-bound ER has emerged, G protein-coupled receptor 30 (GPR 30), which mediates estrogenic responses in cardiovascular and metabolic regulation (Figure 1B). 49 G protein-coupled receptor 30 was identified as the receptor mediating estrogen-induced activation of extracellular signal-regulated kinase 1 and 2 (ERK1/2). More recently, GPR 30 has been shown to activate rapid signaling cascades, such as PI3K, after estrogen binding. G protein-coupled receptor is an intracellular transmembrane GPCR that mediates rapid estrogen signaling. 50 –53 G protein-coupled receptors have both the binding and the signaling functions of membrane ER. 54
Role of eNOS in Cardiovascular System
In the CVS primary endogenous source of NO generation is eNOS. 12,13 Translocation and Akt-dependent phosphorylation at Ser1177 are 2 major ways of eNOS activation. 55 –57 Endothelial NOS protein possesses multiple putative phosphorylation sites, which can be phosphorylated by various protein kinases. 58 –60 Phosphatidylinositol 3-kinase/Akt signaling cascade is capable of directly activating eNOS via phosphorylation at Ser1177. 55,61 The vasodilatory activity of NO occurs in the VSMC due to its interaction with the iron heme in sGC. 13,62 This interaction activates the enzyme to produce cGMP from guanosine 5′-triphosphate. 13,63 Soluble guanylyl cyclase increases cGMP and also stimulates cGMP-dependent protein kinases (cGK type I and type II). 7,64 –66 In CVS, the predominantly expressed type is cGKI. 7,64 –66 Elevated levels of NO and cGMP can attenuate Ca2+ influx and start Ca2+ removal mechanisms, decreasing the endothelial [Ca2+]i in a negative-feedback fashion. 67
Fluid shear stress and numerous agonists are activators eNOS, via cellular events. 68 A host of mechanisms including increased intracellular Ca2+, interaction with substrate and cofactors, activators of protein phosphorylation, and others receptor-mediated agonists may lead to eNOS activation. 68 Dysregulation of these processes results in reduced NO synthesis and release, as a result of attenuated eNOS activity. 68 Nitric oxide impacts platelet function, 13,69 angiogenesis, 13,70,71 cellular respiration, 13,72 and protection from ischemia reperfusion injury. 13,73,74 Endothelial NOS is an important modulator of angiogenesis and vascular tone. 59,75
Nitric oxide is known to interfere with tyrosine phosphorylation and can either diminish the efficacy of a protein as a substrate for tyrosine kinases or lead to their activation by increased phosphorylation directly 76 –78 or by inactivation of phosphatases. 79 The production of large amounts of NO during inflammatory challenge leads to the formation of reactive nitrogen species/reactive oxygen species capable of oxidizing many biological molecules including protein tyrosine nitration. 80 A series of studies demonstrated protein nitration in the human atherosclerotic tissue. 80 Protein tyrosine nitration was associated with iNOS expression and detected in iNOS-positive macrophage-rich lesions at different stages of atherosclerosis. 80 –82 Presumably, tyrosine nitrated proteins with altered function may promote atherogenesis, counteracting the well-established antiatherogenic effects of NO. 80
Also, the results of cross-sectional study suggested that posttranslational modification of proteins via nitration within atherosclerotic plaque-laden arteries and in circulation serve as neoepitopes for the elaboration of immunoglobulins thereby providing an association between oxidant production and the activation of the immune system in coronary artery disease. 83
Regulation of eNOS by Estrogens
Considerable evidence exists suggesting that estradiol plays a cardioprotective role in women’s vascular health.
84,85
The molecular mechanisms by which estradiol regulates eNOS expression, activity, and function is an area of intense interest and requires further research.
86
The eNOS activation by estrogen leads to improvement in vascular health and also reduces cardiomyocyte death.
87
The NOS is present in vascular ECs as a multidomain enzyme consisting of an N-terminal oxygenase domain (amino acids 1-491) that contains binding sites for heme,
Interest in this pathway increased substantially in 1997, when Lantin-Hermoso et al 93 and Caulin-Glaser et al 94 reported that relatively specific antagonists of the ERs could inhibit NO release from cultured EC exposed to physiologic concentrations of estradiol. 86
It has been shown that palmitoylation of ERα regulates its localization to the plasma membrane, where it is localizes in caveolae. 4,45,95,96 Caveolae are invaginations in the cell membrane known to contain eNOS and other signaling proteins. 86 Caveolin 1 is a caveola structural protein that directs caveolae targeting of multiple signaling molecules. 97 –99 Estrogen receptor α interacts with caveolin 1, c-Src, Akt, PI3K, heat shock protein 90 (HSP90), and eNOS forming a functioning complex in caveolae. 4,100,101 Estrogen receptor α's colocalization with HSP90 and eNOS within the caveolae facilitates activation of eNOS by the estradiol. 4,45,95,102 Estradiol binding to ERα localized to the plasma membrane produces steroid receptor fast-action complex in caveolae thus eliciting Gαi activation, which mediates downstream events. 4 The chaperone protein, HSP90, facilitates interaction between the ER and eNOS and other signaling molecules. 4 The increased synthesis and release of NO in estrogen target tissue is attributed to estradiol activation of ER and the latter upregulating eNOS expression and activity via specific phosphorylation. 87
In EC, estradiol activates eNOS via interaction with its receptor; however, whether both ERα and ERβ activate eNOS or specific receptor subtypes may have differential ability to activate eNOS remains to be investigated. 4 Also, ERα and ERβ are present in VSMCs and localize predominantly in the nucleus. 4 However, both receptors are thought to be present at the plasma membrane and associate with caveolin 1. 4 Some studies suggested that there is more ERα than ERβ present in adult VSMCs. 4 In cardiac myocytes, the data on the intracellular distribution of the ER isoforms is at best rudimentary. 4
In 2000, two laboratories provided substantial evidence for the involvement of a signal transduction pathway in eNOS activation: PI3K/Akt pathway. 86,103,104 The activation of Akt mediates many of the downstream cellular effects of PI3K, including activation of cell survival pathways and activation of eNOS. 1,105 Endothelial NOS activation by estradiol can be blocked by cotreatment of EC with a selective PI3K inhibitor, wortmannin, as well as by the transfection of a negative dominant form of PI3K. 1 Main signal-transduction pathway for agonist-stimulated eNOS activation, in EC, depends on Ca2+/CaM/caveolin. 106 Some agonists for NO production need a rise in [Ca2+]. 106 –108 These agonists are acetylcholine, histamine, and bradykinin. 106 –108 In other cases, a rise in [Ca2+]i concentration is not required for NO production. 109 This is characteristic of other forms of stimuli, like estradiol, 94,109 fluid shear stress, 109,110 and insulin/insulin-like growth factor. 109,111
Activation of ERK pathway also has an important role in acute activation of eNOS by estradiol. 58,112 It should be noted that estradiol binding to its membrane receptor results in increased levels of second messengers and activation of a host of signalization pathways including Ca2+, cyclic adenosine monophosphate, NO, and the activation of tyrosine kinase receptors (insulin-like growth factor 1 receptor), epidermal growth factor receptor, Src kinases, protein kinase C, and protein kinase A. 113 –115
The protective effects of estradiol in vascular health are attributed to the genomic and nongenomic actions of estradiol in the vasculature. 3,116 –118 Although considerable details have been delineated, the exact mechanisms of eNOS activation by estradiol remain under intensive investigation. 107
Evidence From Human Studies
Evidence for the importance of eNOS-derived NO in the regulation of vascular tone is based on the experiments in humans 119 in vivo and in vitro. 120
Novensa et al 21 have measured NO, messenger RNA expression, and transcriptional activity by estrogens in human aortic ECs (HAECs). They found an important disparity in the modulation of NO production by estradiol and by conjugated equine estrogens and showed that estrone exhibited a slightly lower potential to increase NO production compared with estradiol, whereas all other estrogens were significantly less effective. 21 Human aortic ECs were loaded with diaminofluorescein 2 diacetate and excited with light at 495 nm, and weak green fluorescence was observed under basal conditions. 21 Treatment with estradiol and estrone caused a significant dose-dependent, saturable increase in green fluorescence indicating an increase in intracellular NO. 21 Estradiol induced a dose-dependent increase in eNOS activity. 21
Role of estradiol was also observed in human umbilical artery endothelial cells that were isolated from arteries of fresh human umbilical cords. 119 Estradiol increased eNOS phosphorylation on Ser1177/total eNOS ratio. 119
In human umbilical vein endotelial cells (HUVEC), both ERα and ERβ are expressed, and estradiol potentiates endothelium–dependent, flow-mediated vasodilatation in the brachial artery of postmenopausal women. 121,122 In addition, in vitro study on HUVECs show that combination of estradiol and a NOS inhibitor inhibited NO increase, indicating that NOS is involved in the estradiol-mediated enzymatic mechanism to increase NO. 123
Scarabin-Carre et al conducted the first study that shows a positive association between plasma estradiol levels and the risk of both coronary heart disease (CHD) and ischemic stroke among postmenopausal women >65 years of age. These associations were independent of traditional cardiovascular risk factors such as diabetes. 124
The Women's Interagency HIV Study showed that HIV-infected women had reduced estrogen and androgen compared with uninfected premenopausal women. Estradiol deficiency is linked with carotid stiffness among immunocompromised HIV-infected premenopausal women. 125 However, reports from randomized clinical trials such as the Heart and Estrogen/Progestin Replacement Study and the Women's Health Initiative, which examined the effects of conjugated equine estrogens in older women with CVD, denied the protective vascular effects of estrogen treatment. 126 Additional data from other studies have supported the concept that the vasoprotective effects of estrogen are evident only when hormone treatment is initiated soon after the onset of menopause and before the development of atherosclerosis. 17
On the other hand, it has been reported that using oral contraceptives containing chlormadinone and ethinylestradiol reduces mean carotid artery stiffness by 12.5% after 12 months of follow-up. 127
Some of the effects of estradiol generated from human studies are summarized in Table 1.
Effects of Estradiol: Evidence From Human Studies.
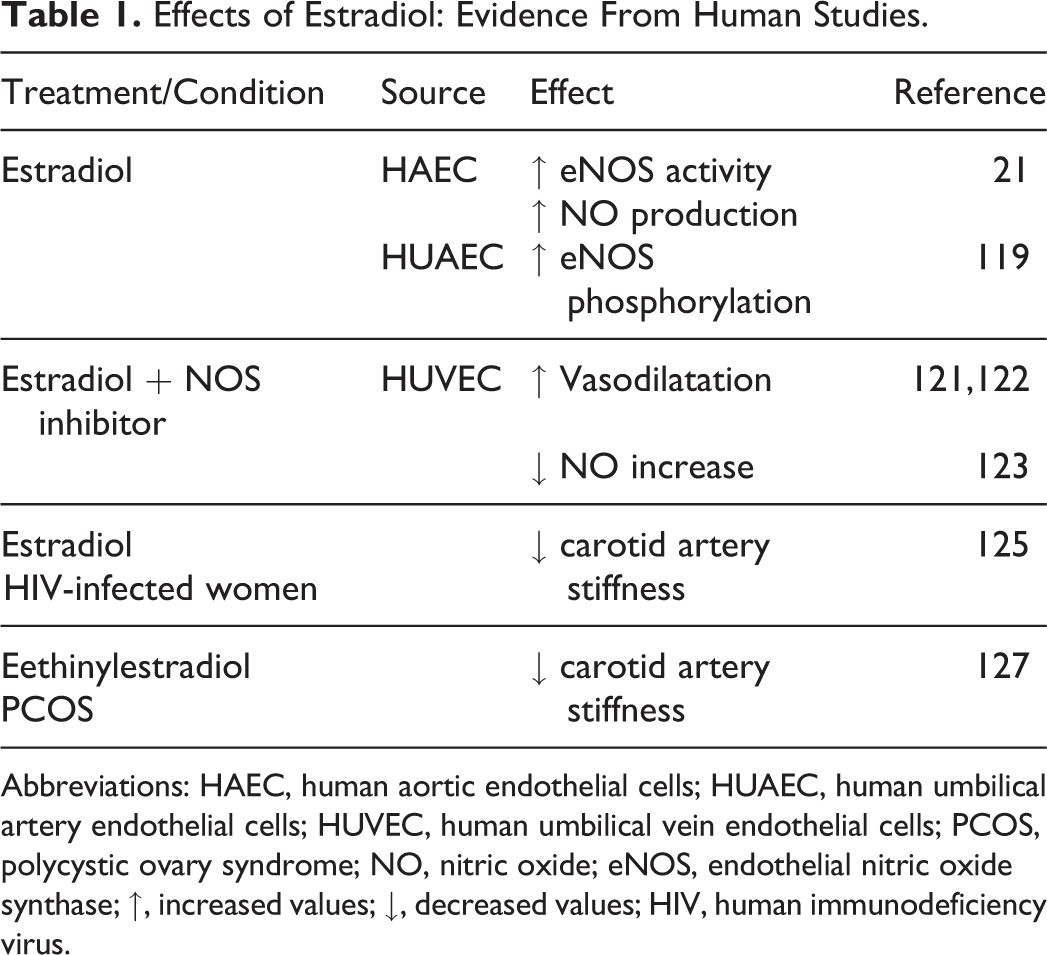
Abbreviations: HAEC, human aortic endothelial cells; HUAEC, human umbilical artery endothelial cells; HUVEC, human umbilical vein endothelial cells; PCOS, polycystic ovary syndrome; NO, nitric oxide; eNOS, endothelial nitric oxide synthase; ↑, increased values; ↓, decreased values; HIV, human immunodeficiency virus.
Evidence From Animal Studies
In 1997, Lantin-Hermoso et al 93 demonstrated that eNOS activity increased in a dose-dependent manner within 5 minutes after ovine pulmonary artery EC were exposed to estradiol. 86 In their studies, 93 removal of ionic Ca2+ completely inhibited estradiol–stimulated eNOS activity, supporting the hypothesis that estradiol acutely stimulates eNOS by increasing Ca2+ influx. 86
We have also studied regulation of cardiac eNOS by estradiol, and our results show that estradiol increased eNOS phosphorylation in rat hearts 22 and in rat aortic ECs in vivo. 128
Endothelium-dependent vascular relaxation is greater in female than male spontaneously hypertensive rats (SHR), and also ERα agonists improve EC dysfunction in blood vessels of ovariectomized (OVX) female SHR. 121,129 Nitric oxide production and estradiol-induced vascular relaxation are greater in mice expressing only ERα. 121,130,131 Estradiol administration in OVX female mice caused rapid arterial dilation of elastic and muscular arteries and ER-mediated NO production. Estradiol causes rapid activation of mitogen-activated protein kinases/ERK and PI3K activity in these arteries. Estradiol-induced kinase activation and vasodilator responses are absent in both ERα and ERβ knockout mice, implicating both ER subtypes in mediating estradiol actions. Thus, estradiol modulation of arterial tone in vivo may involve plasma membrane ER and rapid signaling. 25,121
Vascular contraction is greater in blood vessels of male than female rats, no different between castrated and intact males, and greater in OVX than control females. Estrogen replacement in OVX female rats restores vascular contractility, suggesting that the gender differences in vascular contraction may involve direct effects of estrogen on the vasculature. 132
Effects of estradiol were also described in arteries from male rats. 133 Observations by Huang et al 134 showed that estradiol restored endothelial NO release in response to shear stress in pressurized gracilis muscle arterioles of male SHR by upregulation of eNOS. Earlier reports indicated that basal release of NO is increased in females compared to males, 133,135,136 and that estradiol administration to OVX rats restores the impaired ex vivo basal release of NO. 133 In aortas from a female mouse model of type 2 diabetes (vs nondiabetic controls), Taguchi et al 116 found that estradiol-induced vascular relaxation and NO production were markedly increased and that eNOS phosphorylation under estradiol stimulation was not decreased; actually, it was increased. 116 Another study 123 on rats shows in vivo a positive effect of application of estradiol on tissue survival and activation of eNOS resulting in elevated NO levels. 123
Han et al 137 showed with staining that the vascular wall of rat aortas tended to be thicker in OVX groups. They also found that phosphorylation of eNOS increased after estradiol stimulation. 137 Another study on rats found that serum triglyceride (TC) and low-density lipoprotein cholesterol (LDL-C) levels were increased in high-fat diet (HFD)-fed OVX rats. Administration of estradiol in OVX plus HFD rats could significantly decrease serum TC and LDL-C levels. 138
Protective influence of estradiol and ERα agonist 16a-LE2 on the heart, under conditions of pressure overload, has been shown on mice. Both substances decelerated the hypertrophic response of the left ventricle and prevented the loss of cardiac function. Thus, the protective effects of estradiol and 16a-LE2 are also reflected by reduced fibrosis formation. 139
Histological examination of the thoracic aortas of rabbits revealed that the area of atherosclerosis in the thoracic aorta was reduced by 70% in the cholesterol-enriched diet estradiol-treated group compared with the cholesterol-enriched diet group receiving placebo pellets. 140
Some of effects of estradiol generated from animal studies are summarized in Table 2.
Effects of Estradiol: Evidence From Animal Studies.a

Abbreviations: eNOS, endothelial nitric oxide synthase; NO, nitric oxide; Ca2+, calcium ion; RAEC, rat aortic endothelial cells; OVX, ovariectomized; SHR, spontaneously hypertensive rats; T2D, diabetes type 2; HFD, high fat diet; HCD, cholesterol-enriched diet; LDL-C, low-density lipoprotein-cholesterol; TC, total cholesterol; ↑, increased values; ↓, decreased values; ERα, estrogen receptor α.
Estrogens and eNOS in HDL Functionality and Atheroprotection
A strong association between HDL cholesterol (HDL-C) levels and the risk of developing atherosclerotic vascular disease has been frequently reported. 141,142 High-density lipoprotein has been shown to have a variety of atheroprotective properties, including reverse cholesterol transport and protection from lipoprotein oxidation. 141,143 –145 Also, several studies have indicated that HDL stimulates EC NO production 146 –149 and promotes endothelial repair mechanisms. 150 –155 Therefore, HDL-mediated NO generation via phosphorylation of eNOS at residue Ser1177 and Thr495 seems to be an important factor for regulation of endothelial function. 156 These protective effects of HDL on eNOS and endothelial repair are, at least in part, mediated via endothelial scavenger receptor B, type I (SR-BI). 146,147,150,151 Moreover, by binding to SR-BI, HDL has been reported to activate eNOS via phosphorylation of eNOS at Ser1177, through PI3K and Akt kinase. 147,150
High-density lipoprotein is a lipoprotein class that can be found in either a discoidal (rich in free cholesterol) or spherical (rich in cholesteryl esters) form, according to its lipid content. 157 In addition to the traditional view that HDL-C levels in plasma are associated directly with atheroprotection, recent data indicated that HDL particle functionality may also be an important parameter for atheroprotection. 158,159 In particular, these studies suggested that HDL particle functionality depends on its apoprotein and lipid composition.
Some of the latest proteomic studies 160,161 have identified up to 75 distinct proteins associated with centrifugally isolated HDL raising the possibility that apolipoprotein (Apo) composition may determine particle functionality. 162,163 Although traditionally HDL is referred to as a single particle that carries all HDL-associated Apos, results from nondenaturing 2-dimensional electrophoresis analysis revealed the presence of HDL subpopulations in the same individual with distinct apoprotein and lipid composition. 164 Given that only last year it became obvious that an HDL particle can accommodate more than 2 ApoA-I molecules 165 as it was originally proposed, 166 we still need to learn more about HDL biochemistry and pharmacology. To this date however, it is well accepted that the main atheroprotective properties of HDL are the reverse cholesterol transport where HDL gathers excess cholesterol form peripheral tissues, 161,167 the prevention of LDL oxidation, 168 the protection against inflammation, 169 and the stimulation of eNOS. 148 It is possible that the ability of HDL to stimulate eNOS production may be due at least in part in estrogens present in HDL.
Indeed, research by Kanji et al 170 showed that estradiol, which is the most biologically potent estrogen, associates with HDL in the form of fatty acyl esters that are generated by the plasma enzyme lecithin-cholesterol acyl transferase. 170 Thus, in addition to the classical lipids (cholesterol, cholesteryl esters, and TCs), recent data indicated that fatty acyl esters of estrogen may also be present in the lipid core of an HDL particle. More recent studies showed that HDL-associated estradiol esters contribute indirectly to the atheroprotective potential of HDL by stimulating the enzymatic activity of eNOS, thus leading to the production of NO. Observational studies and clinical trials of estradiol use have shown that estradiol favorably alter the lipoprotein profile, by decreasing LDL and ApoB and increasing HDL and TCs, primarily by influencing the expression of hepatic apoprotein genes. 17,171 Other studies in cultures of human microvascular EC showed that HDL isolated from female patients was capable of inducing eNOS activity in an SR-BI-dependent manner, while HDL from men had minimal activity. 172 Of note, HDL from female patients was enriched in estrogen esters compared to male patients. This SR-BI dependence raises the possibility that in addition to mediating reverse cholesterol transport, binding of HDL to SR-BI may also signal for the increase in vascular eNOS activity. In line with this, only HDL isolated from female patients was capable of promoting vasorelaxation in arterial relaxation studies. 172 The same applies to experimental mice, where only female and not male HDL promotes muscle relaxation. Other in vitro studies further suggested that in addition to SR-BI, the LDL may also play important role in the uptake of estradiol containing HDL. 173 Also, cholesterol efflux assays using cholesterol-loaded THP-1 macrophages and HDL-associated estradiol esters or with HDL isolated from premenopausal females and males of the same age, indicated that estradiol-associated HDL significantly increased cholesterol efflux. Importantly, impairment of either SR-BI or ER markedly reduced cholesterol efflux in these cells. 174 Overall these studies suggest that HDL serves as a vehicle for estradiol and leads to an additional module of HDL-mediated atheroprotection via stimulation of eNOS and production of vascular NO.
In a Study of Women Across the Nation (SWAN ancillary study) comparing subclinical atherosclerosis in premenopausal/early premenopausal with late premenopausal/postmenopausal women not using hormone therapy, aortic calcification, coronary artery calcium, and intimal medial thickness were measured and results show that the protective effects of HDL-C may be diminished during the menopause transition. 175,176
HDL exhibits potent cardioprotective, antioxidant, and anti-inflammatory properties. The antinflammatory properties are partly due to a reduction in smooth muscle cells chemokine expression and proliferation via pERK inhibition, and SR-BI appears to play a crucial role in mediating these effects 177 . Wu et al. reported that discoidal reconstituted HDL containing ApoA-I complexed with phosphatidylcholine inhibit vascular cell adhesion molecule-1, intercellular adhesion molecule-1 and E-selectin expression in tumor necrosis factor α -induced inflammation in cultured human ECs in an SR-BI -dependent manner 178 .
Basler et al. suggests that in contrast to healthy subjects, in patients with coronary artery disease or acute coronary syndrome HDL fails to stimulate NO production and eNOS-activating pathways 150 . This attenuation in the capacity of HDL to stimulate the production of NO may reflect changes in HDL functionality that in turn render it incapable of promoting endothelial repair in the artery 150 .
Conclusions
Previous investigations have documented the critical role of estradiol on the regulation of NOS. Disruption of expression, activity, or functions of eNOS due to any pathology will lead to the development of CVD. The protective effect of estradiol on the development of CVD may serve as a basis for developing new pharmacotherapeutic drugs to help ameliorate the disease and reduce deaths caused by CVD. In this review, we presented contemporary data from recent animal and human studies, which focused on estradiol actions on cardiovascular physiology and pathology. Analysis of the pertinent literature points to possible protective effects of estradiol on the vasculature and may reduce the incidence of CVD.
Footnotes
Author’s Note
This work is part of collaboration between University of St Andrews, School of Medicine, St Andrews, United Kingdom, Department of Medicine, Pharmacology Unit, University of Patras Medical School, Panepistimioupolis, Rio, Greece, and Institute Vinca, Serbia.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work is supported by the grant no. 173033 (to ERI) from the Ministry of Science, Republic of Serbia.