
Abstract
Purpose:
To evaluate whether evacetrapib prolongs QT intervals in healthy participants.
Methods:
This was a single-center, randomized, active and placebo-controlled, 3-period, 6-sequence, and crossover study. Participants were randomized to 1 of 6 treatment sequences in which they received 1 of 3 treatments: evacetrapib 1200 mg daily for 10 days (supratherapeutic dose), moxifloxacin 400 mg for 1 day (positive control), or placebo for 10 days in each of the 3 separate treatment periods. Electrocardiographic parameters were recorded at time points specified in the protocol. The primary end point was the comparison of evacetrapib effect on the population-corrected QT interval (QTcP) to that of placebo at 7 time points following dosing on day 10. An upper limit of the 2-sided 90% confidence interval (CI) <10 milliseconds confirmed the absence of significant effect. Pharmacokinetic parameters were also calculated.
Results:
Subjects were predominantly male (73.2%) with a mean age of 43.1 years and a mean body mass index of 25.9 kg/m2. For the primary analysis, the upper bound of the 2-sided 90% CI for the mean difference between evacetrapib and placebo was <10 milliseconds at all time points on day 10. Following administration of moxifloxacin, the QTcP increased by ≥5 milliseconds at all time points (2, 3, and 4 hours postdose). Maximum plasma concentrations of evacetrapib occurred at a median time of approximately 2 hours, and the mean apparent elimination half-life was approximately 41 hours. The area under the curve and Cmax achieved in this study were both ∼5-fold the values that are expected with the dose level being studied in a phase 3 cardiovascular outcome study. A 1200-mg supratherapeutic dose of evacetrapib was considered to be well tolerated after 10 days of daily dosing in healthy participants.
Conclusions:
Evacetrapib is not associated with QT interval prolongation, even at supratherapeutic doses.
Introduction
Evacetrapib is a potent and selective inhibitor of cholesteryl ester transfer protein (CETP) and is currently being investigated as a treatment, in addition to standard of care, to increase high-density lipoprotein cholesterol (HDL-C) levels and decrease low-density lipoprotein cholesterol (LDL-C) levels. Aggressive LDL-C lowering has demonstrated 20% to 35% relative reduction in cardiovascular events; however, there remains a need for additional therapeutic options capable of addressing the residual cardiovascular risk/disease. Epidemiologic evidence indicates that HDL-C levels are inversely correlated with cardiovascular disease risk. 1–3 Inhibition of CETP is one mechanism for increasing HDL-C and lowering LDL-C concentrations, and therefore evacetrapib may address an unmet need.
Evacetrapib is currently in development for the prevention of cardiovascular events, and it is possible that subjects receiving evacetrapib in the future may concurrently take medications that could increase the QT interval. QT prolongation increases the risk of potentially life-threatening ventricular tachyarrhythmias, such as torsades de pointes. 4 Therefore, assessment of the effects of evacetrapib on the QT interval is warranted. Additionally, evaluating QT/QTc early in drug development serves to establish the potential threshold for QT prolongation by evacetrapib and informs later development regarding the intensity of monitoring QT/QTc interval in the targeted population. The CETP inhibitor, dalcetrapib, which was terminated for futility, 5 had been evaluated at above-mentioned therapeutic levels and was not associated with prolongation in a thorough QT (TQT)/QTc. 6 To our knowledge, TQT/QTc data with anacetrapib, a CETP inhibitor in late stage development, have not been disclosed.
Thorough QT studies are designed to determine the potential of drugs to delay cardiac repolarization. Although prior QT data from phase 1 studies with evacetrapib did not reveal any clinically relevant QT findings, this is the first TQT study for evacetrapib addressing regulatory requirements. This TQT study evaluated the effect of multiple once-daily (QD) doses of evacetrapib, at supratherapeutic levels, on the QT interval. Secondary objectives included the evaluation of the safety, tolerability, and pharmacokinetic (PK) profile of evacetrapib when given as multiple QD doses to achieve supratherapeutic exposure. The study design conformed to International Conference on Harmonization (ICH) guidelines for clinical evaluation of QT/QTc interval prolongation. 7,8
Methods and Materials
Study Design
This was a randomized, 3-period, 6-sequence, crossover study designed to investigate the effect of evacetrapib on QT interval corrected for heart rate (QTc). 9 Evacetrapib was administered as multiple QD doses to achieve a supratherapeutic exposure and was designed to conform with International Conference on Harmonisation (ICH) guidelines for a TQT/QTc study. 7,8 The trial was placebo controlled and included a moxifloxacin-treated group as a positive control, as the fluoroquinolone antibiotic is known to prolong the QT interval by an average of 7.5 to 12.8 milliseconds. 10 Subjects were randomized to 1 of the 6 treatment sequences in which they received 1 of the 3 treatments: (1) evacetrapib, 1200 mg/day (supratherapeutic dose) for 10 days, (2) placebo for 10 days, or (3) moxifloxacin, 400 mg for 1 day, in each of 3 separate treatment periods (Figure 1). The dose selection for this TQT study was supported by a previously conducted phase 1, multiple-dose study in healthy adults which demonstrated evacetrapib 1200 mg daily for 14 days to be well tolerated (Eli Lilly, data on file). The subjects and investigators were blinded to the evacetrapib and placebo treatments; however, the moxifloxacin was given unblinded.

Study design. Treatment A = days 1 to 10 evacetrapib 1200 mg/day, treatment B = days 1 to 10 placebo (matching evacetrapib in size and appearance), and treatment C = day 1 moxifloxacin 400 mg/day. Shaded boxes indicate washout period of ≥14 days. R indicates randomization; ECG, electrocardiogram.
For each subject, the study consisted of a screening visit, 3 dosing periods, and a poststudy follow-up examination. All subjects were admitted to a clinical research unit (CRU) on day −2 of each treatment period. Subjects who received single doses of moxifloxacin were discharged from the CRU after the completion of study procedures at the 24-hour time point on day 2. Subjects who received daily doses of evacetrapib or placebo for 10 days were discharged from the CRU after the completion of study procedures at the 24-hour time point on day 11. There were at least 14 days between consecutive dosing periods. Moxifloxacin, evacetrapib, and placebo were administered orally in the fasted state on the morning of the dosing day in order to obviate food effects that can confound QT measurements.
Study Subjects
Eligible subjects included healthy men and women between 19 and 65 years of age (inclusive), with a body mass index (BMI) of 18.5 to 29 kg/m2. Subjects were excluded from participation if they had any electrocardiographic (ECG) abnormality that may increase the risk associated with participating in the study or confound the QTc analysis. Subjects were additionally excluded from participation if they had a personal or family history of long QT syndrome, heart failure, hypokalemia, family history of sudden death, or personal history of unexplained syncope. Use of over-the-counter or prescription medication was prohibited within 14 days prior to dosing or during the study.
The study protocol was approved by an institutional review board and was conducted in accordance with the Declaration of Helsinki and Good Clinical Practice guidelines. All subjects provided written informed consent prior to participating.
Electrocardiographic Assessments
Electrocardiographic assessment for evaluation of changes in the QT interval was performed by continuous ambulatory 12-lead ECG recordings on days −1, 1, 2, 10, 11, 13, 15, and 17 during the evacetrapib and placebo dosing periods and on days −1, 1, and 2 during the moxifloxacin dosing period. Subjects were required to remain semisupine for approximately 6 hours postdose and were instructed to minimize activity and environment-induced heart rate perturbations. During the day −1 collection, 1 time point was collected in a standing position in order to raise the subject's heart rate. For each nominal time point, 5 unique replicates (quintuplicate) ECGs with 1 minute between each replicate were selected, and QT intervals were measured. 11 Measurement of QT was performed on superimposed median waveforms from all 12 ECG leads. A central ECG laboratory conducted a full overread on 1 of the replicate ECGs (including all intervals).
Pharmacokinetic Assessments
Blood samples were collected for PK analysis predose and at 1, 2, 3, 4, 6, 12, 24, 72, 120, and 168 hours following the evacetrapib or placebo dose on day 10 of each respective dosing period. Plasma samples were analyzed at BASI (West Lafayette, Indiana). Samples were analyzed for evacetrapib using a validated liquid chromatography with tandem mass spectrometry method. The lower limit of quantification was 1.00 ng/mL, and the upper limit of quantification was 1000 ng/mL.
Safety Assessments
Safety was assessed by physical examinations, clinical safety laboratory tests, vital signs, and safety ECGs as well as the monitoring of treatment emergent adverse events (AEs). Assessment of clinical laboratory safety parameters included hematology, biochemistry, urinalysis, and coagulation parameters. A skin and targeted examination based on reported symptoms occurred at days 1, 2, 10, and at the follow-up visit (following the evacetrapib and placebo dosing periods).
Statistical Analysis
The individual-corrected QT interval (QTcI), population-corrected QT interval (QTcP), and Fridericia-corrected QT interval (QTcF) were calculated for each replicate ECG measurement. The QTcP produced the slope closest to 0 in the regression of QTc versus the onset of 1 QRS complex to the onset of the next QRS complex (RR), indicating that a positive correlation was minimized and was therefore selected as the primary variable. Population-corrected QT interval was calculated as follows: QTcP = QT/RRβ, where β is the population correction factor computed from a log-linear model ln QT = α + β × ln, RR fitted to all QT and RR measurements on day 1 of period 1 and predose on day 1 in each period for all subjects. For each period, the average of 5 predose time point (−2, −1.5, −1, −0.5, and 0 hours) QTc measurements on day 1 was considered the baseline QTc. The average of the quintuplicate (5 replicate ECGs approximately 1 minute apart) QTc measurements at each time point following dosing on day 10 was calculated for each subject and for each period.
The primary statistical analysis was the comparison of evacetrapib effect on QTcP to that of placebo at 1, 2, 3, 4, 6, 12, and 24 hours following dosing on day 10 using a linear mixed-effects model. Baseline QTc values were included as a covariate. Treatment (evacetrapib and placebo), time, period, sequence, and the time-by-treatment interaction were included as fixed effects. Subject and subject-by-period interaction were included as random effects. Moxifloxacin’s effect on QTc interval was compared to that of placebo using a linear mixed-effects model similar to that used in the statistical analysis of evacetrapib. To establish sensitivity, the lower bound of the 90% confidence interval (CI) for the difference between moxifloxacin and placebo groups must be ≥5 milliseconds at ≥1 time point.
It was calculated that 52 subjects were needed to complete the study in order to provide approximately 90% probability that the upper limit of the 2-sided 90% CI (equivalent to the upper 1-sided 95% CI) for the baseline-adjusted mean difference between evacetrapib and placebo is <10 milliseconds. An intrapratient standard deviation of 5.44 milliseconds and a subject-by-day-by-time standard deviation of 3.13 milliseconds for QTcF, based on the ECG measurement in males and females, were assumed in the calculation of the sample size. 12
For the PK analysis, the area under the concentration versus time curve during 1 dosing interval at steady state (AUCτ, ss); maximum observed drug concentration during a dosing interval at steady state (Cmax, ss); and other PK parameters were determined from the plasma concentrations of evacetrapib, using noncompartmental procedures (WinNonlin Version 5.2; Pharsight Corporation, Mountain View, California).
Results
In total, 71 healthy participants participated in the study and received at least 1 dose of evacetrapib or placebo. Sixty-six subjects completed the study. Of the 5 subjects that did not complete the study, 2 discontinued due to physician decision, 1 discontinued due to personal reason, 2 withdrew consent, and 1 discontinued due to a serious AE (SAE) of deep vein thrombosis.
Demographic characteristics are shown in Table 1. In all, 73.2% of the subjects were male. The mean age was 43.1 (range 19-65) years, and the mean BMI was 25.86 (range 20.3-29.1) kg/m2.
Subject Demographics.

Abbreviations: SD, standard deviation; N, number of subjects studied.
Electrocardiographic Recordings
For the primary analysis of QTcP using a baseline of the predose average measurements on day 1, the upper bound of the 2-sided 90% CI for the mean difference between evacetrapib and placebo was <3 milliseconds at all time points on day 10 (Table 2); far below the regulatory threshold of <10 milliseconds.
Comparison of Mean Change From Baseline in Primary QTcP Between Evacetrapib and Placebo on Day 10.
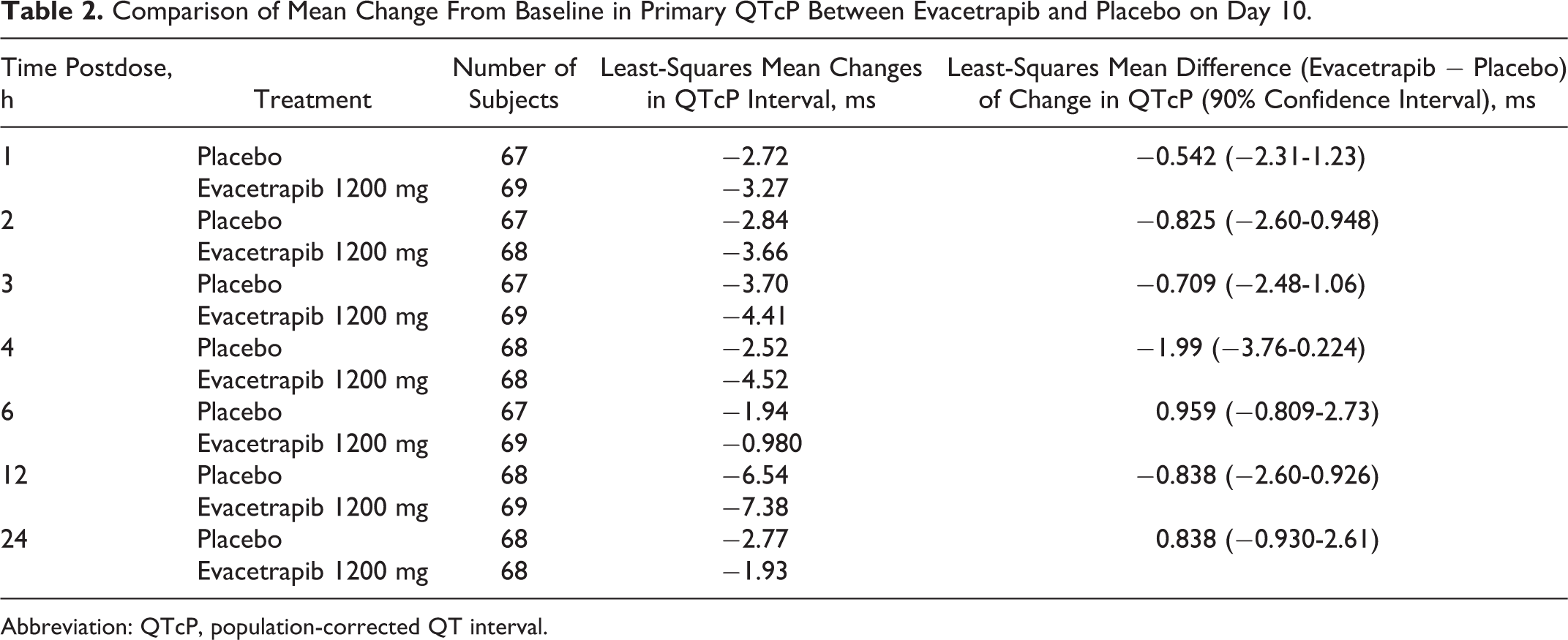
Abbreviation: QTcP, population-corrected QT interval.
A statistical comparison of mean change from baseline (using predose average on day 1) in QTcP between moxifloxacin and placebo is presented in Table 3. The mean change from baseline in QTcP was greater following moxifloxacin compared to placebo at time points on day 1 (least squares [LS] mean differences ranged from 11.9 to 12.5 milliseconds). Assay sensitivity was established in this study as the lower bound of the 90% CI for the difference between moxifloxacin and placebo was ≥5 milliseconds at all time points (2, 3, and 4 hours postdose).
Comparison of Mean Change From Baseline in Primary QTcP Between Moxifloxacin and Placebo on Day 1.

Abbreviation: QTcP, population-corrected QT interval.
As shown in Table 4, no subject experienced a QTcI, QTcF, or QTcP interval prolongation >480 milliseconds (Table 4). Additionally, no subject experienced a >60 milliseconds increase from baseline in 12-lead Holter QTcI, QTcF, or QTcP interval.
Frequency of Absolute 12-Lead Holter QTc Intervals Prolongation.a

Abbreviations: QTcI, individual corrected QT interval; QTcF, Fridericia-corrected QT interval; QTcP, population corrected QT interval; N, number of subjects studied; n, number of subjects.
a Thresholds are based on the mean of quadruplicate measures.
Pharmacokinetics
The mean (SD) evacetrapib plasma concentration–time profile on day 10 of the study following multiple once daily doses of 1200 mg evacetrapib is shown in Figure 2. Maximum plasma concentrations of evacetrapib occurred at a median tmax, ss of approximately 2 hours, ranging from approximately 1 to 6 hours postdose in individual subjects on day 10. Following maximum observed drug concentration (Cmax), plasma concentrations of evacetrapib appeared to decline in a biphasic manner with the start of the apparent terminal elimination phase generally occurring between 12 and 24 hours postdose. The resultant mean apparent elimination half-life was approximately 41 hours, ranging from approximately 26 to 66 hours postdose in individual subjects. The between-subject variability on day 10 was moderate to high with values of 49% and 65% for AUCτ, ss and Cmax, ss, respectively.
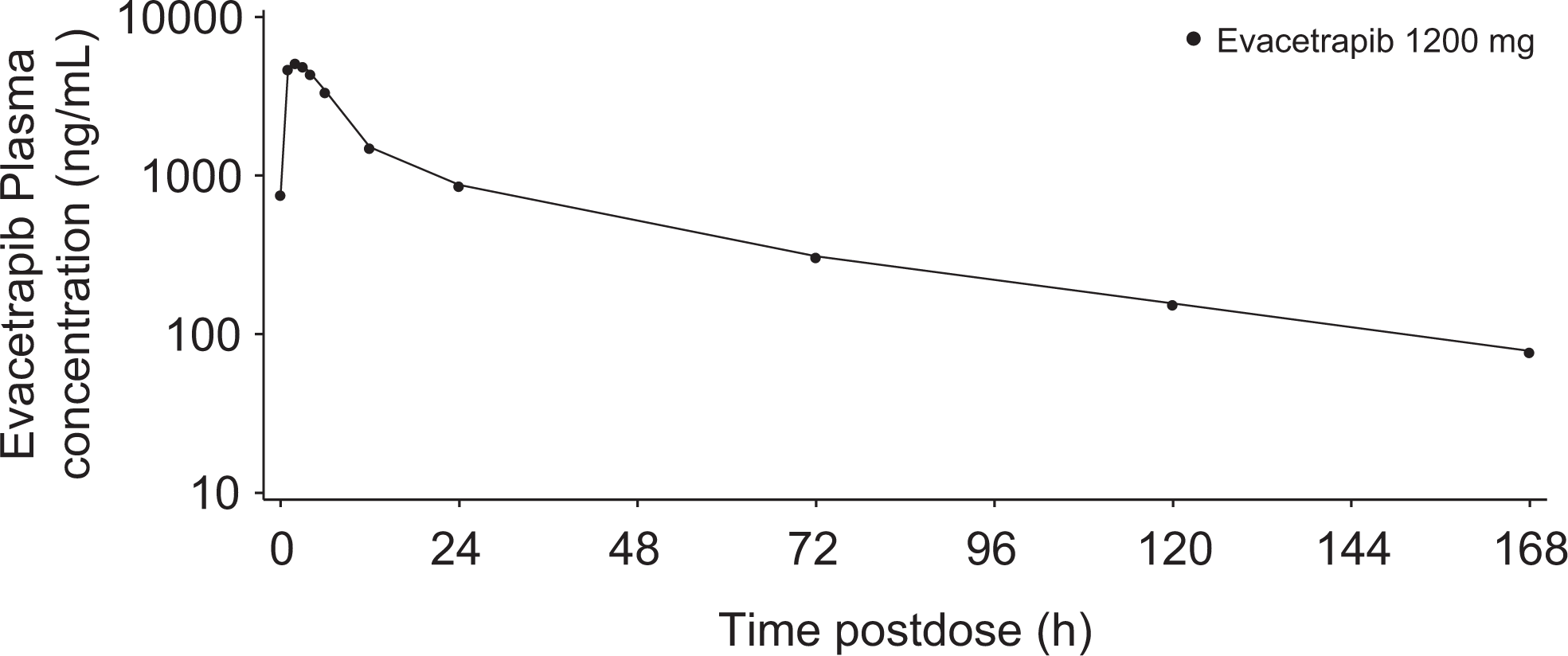
Mean plasma concentrations of evacetrapib following 10 days of once daily administration of 1200 mg evacetrapib. Semilogarithmic scale.
Safety and Tolerability
All AEs reported following the administration of evacetrapib were mild in severity. The most common AEs assigned causality as related to study treatment were under the category of gastrointestinal (GI) disorders (Table 5). The most frequently reported GI disorder was diarrhea, occurring in 20.0% of subjects following evacetrapib treatment and in 5.8% of subjects following placebo treatment. Nausea occurred in 7.1% of subjects following evacetrapib treatment compared with 1.4% of patients following placebo treatment, and abdominal pain occurred in 4.3% of subjects following evacetrapib treatment compared with 0% of subjects following placebo treatment.
Treatment Emergent Adverse Events Related to Study Treatment Occurring in ≥2 Subjects.

Abbreviation: MedDRA, Medical Dictionary for Regulatory Activities.
One SAE of deep vein thrombosis of the left lower extremity was reported during the study during placebo dosing, which was 15 days after the previous dose of evacetrapib. This event resulted in hospitalization, requiring treatment with enoxaparin and warfarin. As a result, the subject was discontinued from the study. This event was considered by the investigator to be related to study treatment.
One AE of a rash on the torso and extremities occurred in a subject approximately 7 hours following the administration of evacetrapib on day 10. The rash was considered mild in severity, was considered related to study treatment, and was resolved spontaneously after approximately 6 days.
One subject had elevated liver enzymes approximately 1 day after the final dose of evacetrapib in period 1. The subject's aspartate aminotransferase (AST) and alanine aminotransferase (ALT) were within normal limits on day 1 and increased ∼2 times the upper limit of normal 1 day following the last dose of evacetrapib. Both the AST and ALT results decreased thereafter and were within the normal reference range by 5 to 7 days after the last dose of evacetrapib. This subject's total bilirubin levels remained within normal limits.
There were no clinically significant findings in the safety assessments from the 12-lead ECGs for individual subjects following administration of evacetrapib, moxifloxacin, or placebo.
Discussion
This TQT study was performed in healthy participants to determine whether evacetrapib delays cardiac repolarization. This study was conducted in accordance with ICH guidelines for the clinical evaluation of QT/QTc interval prolongation. Ten daily supratherapeutic doses of evacetrapib achieved a substantial exposure above the clinical dose, which was not associated with prolongation of the QT interval, as indicated by the largest bounds of the upper 1-sided 95% CI for QTcP being consistently well below the 10-millisecond threshold as specified by the ICH guidelines.
We used 3 methods to correct for heart rate, QTcI, QTcP, and QTcF. Population-corrected QT interval was selected as the primary heart rate correction since it minimized confounding by heart rate. In this study, the QTcP results were comparable with those of QTcI and QTcF, with the upper bound of the 2-sided 90% CI for the mean difference between evacetrapib and placebo being <10 milliseconds at all time points and across all 3 methods on day 10. A consistent increase in QTcP interval following administration of moxifloxacin confirmed the required assay sensitivity to detect small increases in QTc.
The implication of this negative TQT/QTc study for the later stage development of evacetrapib is defined in the E14 guidance, in which the collection of baseline and periodic on-therapy ECGs in accordance with the current investigational practices in each therapeutic field is almost always sufficient evaluation during subsequent stages of drug development. 8 As the therapeutic area for evacetrapib is cardiovascular disease, periodic ECGs will be performed in the cardiovascular outcomes study (ClinicalTrials.gov identifier NCT01687998).
In accordance with ICH guidelines, a supratherapeutic dose was tested in this study. The supratherapeutic dose of 1200 mg was selected, as it was found to be safe in a phase 1 multiple-dose study in healthy volunteers assessing this daily dose for 14 days (Eli Lilly, data on file) and was expected to provide supratherapeutic blood levels relative to the 130-mg dose administered in the ongoing phase 3 ACCELERATE study. In this study, the supratherapeutic dose of 1200 mg achieved an AUC of 48 300 ng h/mL and Cmax of 5270 ng/mL at steady state after 10 daily doses. The 130-mg dose of evacetrapib is expected to produce an AUC of 9800 ng h/mL (90% CI: 9000-11 000) and Cmax of 1200 ng/mL (90% CI: 1000-1300) at steady state.
A multiple-dose study design was selected to assess the effect of evacetrapib on QT at steady state. Additionally, the multiple-dose design addressed the potential for a delayed effect on QT, mediated by either the parent compound or a potentially active metabolite that may not be detected after a single dose. Lastly, the multiple-dose design is consistent with TQT studies for another CETP inhibitor previously in clinical development. 6
In accordance with the ICH guidelines, a positive control was used in this study for the purpose of verifying assay sensitivity. The study included moxifloxacin administered as a single dose at 1 of the 3 periods for each subject. The comparison of ECGs collected at the same time points following administration with moxifloxacin versus placebo demonstrated >5 milliseconds for the QT/QTc intervals, confirming the positive control effects of moxifloxacin in this study. Additionally, this study used Williams design 9 in which the sponsor, investigators, and subjects were blinded to the placebo and evacetrapib but were unblinded to the positive control treatment, moxifloxacin. The Williams design is a variance-balanced design in that each pairwise treatment effect is estimated with the same degree of precision. The use of a double-blinded positive control is deemed not essential according to the current regulatory guidance provided that the personnel responsible for overreading the ECGs are blinded to identify all the treatments, time points, and subjects as was the case in this study. 7,8
To reduce intrinsic variability, quintuplicate ECGs for QT and RR interval assessment were obtained by extraction at defined times, including multiple time points prior to the first dose of each period. 11 In a TQT study, baseline correction is recommended due to the potential for large intersubject variability. 13 The baseline for this study was period specific; that is, baseline assessments were collected prior to dosing on the same day of dosing for each study period. A time-matched baseline is usually not necessary for a crossover trial, as the diurnal variability of QTc is accounted for in the crossover design by taking into account the treatment difference within subjects at matching time points from different periods. In fact, adding a baseline day would increase the variability of the treatment difference. 12
Finally, a 1200-mg, supratherapeutic dose of evacetrapib was considered to be well tolerated after 10 days of daily dosing in healthy male and female subjects. Adverse events related to evacetrapib were mild in severity, with GI disorder events being most frequently reported, consistent with the phase 1, multiple-dose study.
This study has potential limitations. The study was conducted with healthy volunteers, rather than the target population, in order to limit exposure to confounding medications. However, the mechanism of action for QT prolongation is not expected to be affected by the disease state of a subject and, as such, evacetrapib is unlikely to delay cardiac repolarization in the target population. Additionally, administration of supratherapeutic doses beyond the 1200-mg dose level was not possible since such doses may result in increased frequency of GI side effects, thus confounding the primary objective of the QT study. However, this study administered a dose that provides an almost 10× multiple to the daily phase 3 dose, and the mean AUC and Cmax achieved in this study were approximately 4.9- and 4.4-fold (respectively) than the values expected to be achieved in phase 3.
Conclusion
This study with TQT assessments demonstrates that evacetrapib is not associated with prolongation of the QTc interval after 10 daily supratherapeutic doses achieving a steady state in healthy participants. The findings of this study support further clinical development in phase 3 studies.
Footnotes
Acknowledgments
The authors wish to acknowledge the investigators and subjects who participated in this study; Malcolm Mitchell of Eli Lilly and Company (Windlesham, Surrey, United Kingdom) for scientific support; Derek Leishman of Eli Lilly and Company (Indianapolis, Indiana) for scientific support and medical review; and Stephanie Brillhart, of inVentiv Health Clinical for providing writing assistance.
Declaration of Conflicting Interests
The author(s) declared a potential conflict of interest (eg, a financial relationship with the commercial organizations or products discussed in this article) as follows: JGS, SF, KAK, and WZ are employees of, and hold stock in, Eli Lilly and Company.
Funding
The author(s) received the following financial support for the research, authorship, and/or publication of this article: This study was funded by Eli Lilly and Company.