
Abstract
Introduction
Coronary artery disease (CAD) is one of the major causes of death throughout the world. 1 Patients with myocardial infarction (MI) due to CAD also have additional diseases such as arterial hypertension, diabetes mellitus, and hypercholesterolemia that can be associated with an increased incidence of MI. Diabetes mellitus is a major risk factor for ischemic heart disease and more than 50% of deaths among the diabetic population are caused by CAD. 1 ,2
During myocardial ischemia, decreased blood supply of the heart can result in MI and in spite of the necessity of reperfusion to restore the normal function of the heart, abrupt reperfusion of an ischemic myocardium produces further damages described as ischemia/reperfusion (I/R) injury. 3,4 A clinically relevant phenomenon, ischemic postconditioning (IPostC), is a novel endogenous mechanism of myocardial protection in which very short cycles of ischemia and reperfusion are applied immediately at the onset of reperfusion and it has a significant cardioprotective effect. 5,6 This intervention has been studied only in normal animals under normal conditions, but not in the presence of comorbidities and risk factors. Thus, evaluating the effectiveness of postconditioning protocols is warranted not only in normal but also in pathological conditions. 6 The effect of postconditioning in diabetes has not been thoroughly studied yet. 7 Moreover, the interaction of diabetes with cardioprotection by IPostC remains unclear.
It has been reported that the mitochondrion plays an important role in I/R injury, cardioprotection, and diabetes. 8,9 Mitochondrial permeability transition pore (mPTP) opens in the early minutes of reperfusion due to oxidative stress, Ca2+ overload, and decreased adenosine triphosphate (ATP) levels. 4,9 Opening of the mPTP is associated with necrosis and apoptosis, 10 and it might be considered a key event from reversible to irreversible cell death. 11 Administration of cyclosporine-A (CsA) at the onset of reperfusion produces cardioprotection against I/R injury in the normal animals by inhibition of the mPTP opening. 3,9,11
It is known that nitric oxide (NO) participates in the regulation of myocardial contractility and contributes to myocardial protection in ischemic pre and postconditioning. 12 It has been shown that NO activity is changed in the diabetic state and reduced basal availability of NO and impairment of endothelial NO-dependent mechanisms due to dysfunction of the normally protective endothelium may also be involved in the pathogenesis of several cardiovascular diseases, including atherosclerosis, hypertension, coronary heart disease, and arterial thrombotic disorders. 13
Due to complex pathophysiology of the diabetes in patients with underlying ischemic heart disease, 7 investigating the interaction of diabetes with I/R injury and cardioprotective mechanisms is of particular interest. Subsequently, reducing the outcomes of CAD using strategies that target I/R injury would be particularly beneficial in this population. Therefore, the aim of the present study was to investigate the effects of IPostC and CsA on NO activity and myocardial injury induced by I/R in the diabetic myocardium.
Materials and Methods
Animals
Male Wistar rats (body weight: 250-320 g) were randomly divided into control (52 rats) and diabetic (64 rats) groups at the beginning of the experiment. The animals were housed in an animal room at 22°C to 24°C with a 12-hour dark and 12-hour light cycle and given free access to food and water. This study was performed in accordance with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH publication No 85-23, revised 1996) and was approved by the local Animal Care Committee.
Chemicals
Streptozotocin, 2,3,5-triphenyltetrazolium chloride (TTC), and CsA were obtained from Sigma-Aldrich (Germany). All chemicals of the Krebs-Henseleit solution were purchased from Merck (Germany).
Induction of Diabetes
Diabetes was induced by a single intraperitoneal (ip) injection of streptozotocin (STZ; 50 mg/kg). Development of the diabetes was confirmed by measuring blood glucose levels using a glucometer device through the sampling of blood with a small scratch to the tail of the rats, 72 hours later. The animals with blood glucose levels higher than 300 mg/dL were considered diabetic, 1,14 and those with blood glucose levels lower than this value were excluded from the experiment. After 8 weeks of induction of the diabetes, the diabetic animals as well as the controls were sacrificed and all the experiments were performed in isolated perfused hearts.
Surgical Preparation and Isolated Heart Perfusion
Surgical preparation was performed as described previously. 10,15,16 All animals were heparinized (500 IU) and anesthetized with a mixture of ketamine (60 mg/kg) and xylazine (10 mg/kg) ip. The hearts were rapidly excised, then mounted on a Langendorff perfusion apparatus, and retrogradely perfused via the aorta with the Krebs-Henseleit solution (in mmol/L: NaCl 118; KCl 4.7; CaCl2 2.5; MgSO4 1.2; NaHCO3 25; KH2PO4 1.2; and glucose 11.1) at a constant perfusion pressure of 75 mm Hg and pH 7.4. The perfusion solution was gassed with a mixture of 95% O2, 5% CO2 at 37°C. Left ventricular pressures and maximum increase or decrease in the left ventricular pressure (±dP/dt) were recorded by a water-filled latex balloon inserted into the left ventricle (LV) through an incision in the left atrium. The balloon volume was adjusted to producing 5 to 10 mm Hg of diastolic pressure in all the experimental groups. Hemodynamic data were digitized by a data acquisition system (PowerLab; ADInstruments, Australia). Coronary flow was measured by timed collection of coronary effluent. The hearts were excluded from the experiment if their baseline left ventricular pressures were lower than 70 mm Hg in the control rats, or they showed severe arrhythmias.
Induction of Regional I/R
At the onset of the experiment, a 5-0 silk thread was placed around left anterior descending (LAD) coronary artery, close to its origin. After the stabilization period of 15 minutes, all hearts were subjected to regional ischemia for 30 minutes followed by reperfusion for 45 minutes. Regional ischemia and reperfusion were induced by occluding and reopening of the LAD, respectively. An immediate fall in the left ventricular pressure at the onset of index ischemia served as evidence of effective coronary occlusion. 17 Ischemic postconditioning in this study was induced by 3 cycles of 30-second reperfusion and ischemia (3 cycles of 30-second R/I) immediately at the onset of the reperfusion.
Experimental Protocol
The animals were divided into 8 groups as follows (Figure 1): (1) control ([C] n = 7), (2) control with IPostC ([C + IPostC] n = 7), (3) control with CsA ([C + CsA] n = 5), (4) control with IPostC plus CsA ([C + IPostC + CsA] n = 6), (5) diabetic ([D] n = 5), (6) diabetic with IPostC ([D + IPostC] n = 7), (7) diabetic with CsA ([D + CsA] n = 5), and (8) diabetics with IPostC plus CsA ([D + IPostC + CsA] n = 6). In groups receiving CsA, 5 minutes before the onset of reperfusion up to 10 minutes of reperfusion, the hearts were perfused with the Krebs-Henseleit solution containing 0.01 mmol/L CsA.

Experimental protocol. C indicates control; C + IPostC, control with ischemic postconditioning; C + CsA, control with cyclosporine-A; C + IPostC + CsA, control with ischemic postconditioning plus cyclosporine-A; D, diabetic; D + IPostC, diabetic with ischemic postconditioning; D + CsA, diabetic with cyclosporine-A; D + IPostC + CsA, diabetics with ischemic postconditioning plus cyclosporine-A; stab, stabilization.
Measurement of Infarct Size and Risk Zone (RZ)
In another series of experiment with a similar grouping (n = 6 each) with a reperfusion period of 60 minutes, infarct sizes (ISs) were identified as described previously. 5,6 At the end of the experiment, the coronary ligature was retightened at the same site and 3 to 4 mL of 0.25% Evans blue dye was infused via the side arm of the aortic cannula into the coronary system to identify the risk zone (RZ) as unstained from the blue, nonischemic part of the myocardium. The hearts were then frozen at −20°C and thereafter cut into thin slices (2 mm) from the apex to the base. The slices were incubated in 1% 2,3,5-TTC in phosphate-buffered solution, pH 7.4 for 10 to 15 minutes at 37°C. The heart slices were immersed in 10% formalin for 24 hours to identify viable myocardium as red stained, while necrotic (infarcted) tissue remains pale gray. The LV, RZ, and IS areas were determined by computerized planimetry (Summa Sketch III, SummaGraphics, USA) and the volumes were calculated by multiplying areas by slice thickness and summing them for each heart. The RZ was expressed as a percentage of the LV (RZ/LV) and the IS was expressed as a percentage of the RZ (IS/RZ).
Measurements of NO
Nitric oxide production in heart homogenates was determined by measuring the total nitrite and nitrate concentration (NO metabolites; nitric oxide [NOx]) using the Griess method. 18 Deproteinized heart homogenate samples were used for NOx determination. Briefly, 100 μL of the supernatant was transferred to a micotiter plate well and 100 μL vanadium (III) chloride (8 mg/mL) was added to each well (for reduction of nitrate to nitrite) and this was followed by the addition of the Griess reagents including 50 μL sulfanilamide (2%) and 50 μL N-(1-naphthyl) ethylendiamine dihydrochloride (0.1%). After 30 minutes of incubation at 37°C, the absorbance was measured in 540 nm using the enzyme-linked immunosorbent assay (ELISA) reader (Lab System, Finland). Concentration of NOx in the heart homogenate samples was determined through a linear standard curve established by 0 to 200 μmol/L sodium nitrite.
Statistical Analysis
All values were expressed as mean ± standard error of the mean (SEM). Hemodynamic data were measured during baseline, ischemia and at 5, 15, 30, and 45 minutes of reperfusion. The hemodynamic parameters were analyzed using repeated measures analysis of variance (ANOVA) with Bonferroni test, and body weight, heart weight, biochemical measurements, and IS were analyzed using one way ANOVA followed by Tukey post hoc test. Differences were considered statistically significant when P < .05.
Results
Characteristics of the Animals
STZ-induced diabetic rats showed a higher mortality rate; 12 diabetic rats died during the 8-week disease period. The rats with a blood glucose level lower than 300 mg/dL after 8 weeks (3 in diabetics), or with lower baseline ventricular pressures or arrhythmias (3 in controls and 2 in diabetics) were excluded from the experiment.
After 8 weeks, the diabetic rats had higher levels of blood glucose as compared to the control animals. Mean values for blood glucose levels of rats are listed in Table 1. There was no difference in body weight between the control and the diabetic animals at the onset of the experiment (269 ± 7 g vs 277 ± 11 g, respectively). However, after 8 weeks, the diabetic rats had a significantly lower body weight (P < .01) and a higher ratio of heart weight to body weight (P < .05), comparing to the controls.
General Information of Control and Diabetic Rats After 8 Weeks a
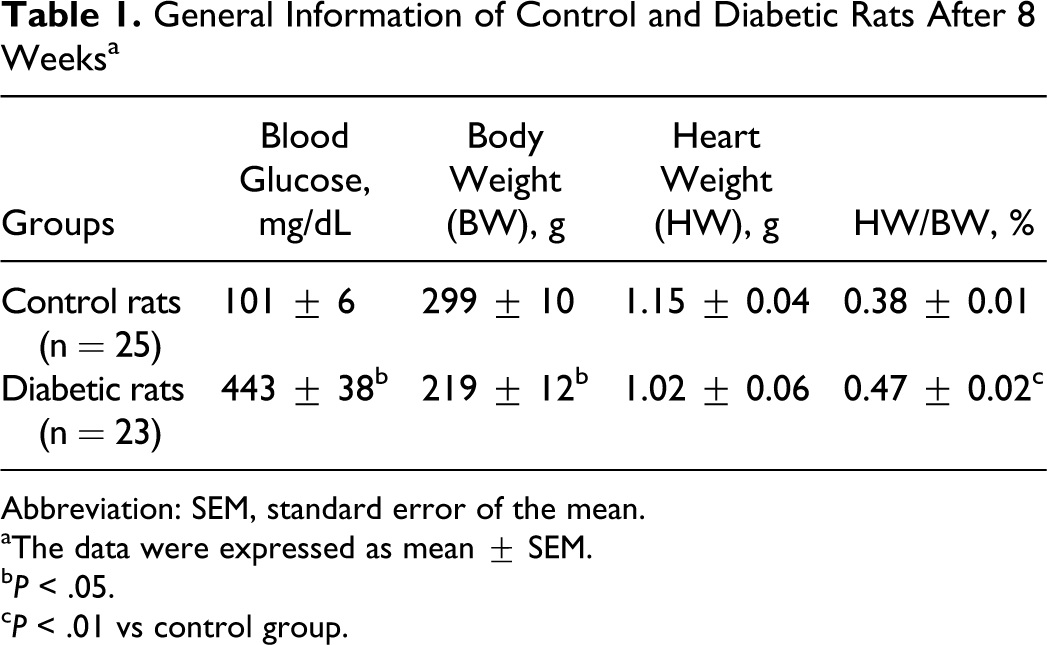
Abbreviation: SEM, standard error of the mean.
aThe data were expressed as mean ± SEM.
b P < .05.
c P < .01 vs control group.
Hemodynamic Parameters
During the preischemic period, the baseline values of hemodynamic parameters (left ventricular developed pressure [LVDP], +dP/dt and −dP/dt) were lower in all the diabetic rats as compared with those in the control animals, indicating the development of myocardial dysfunction in the diabetic rats (Table 2).
Baseline Values of Hemodynamical Parameters in Control and Diabetic Rats a
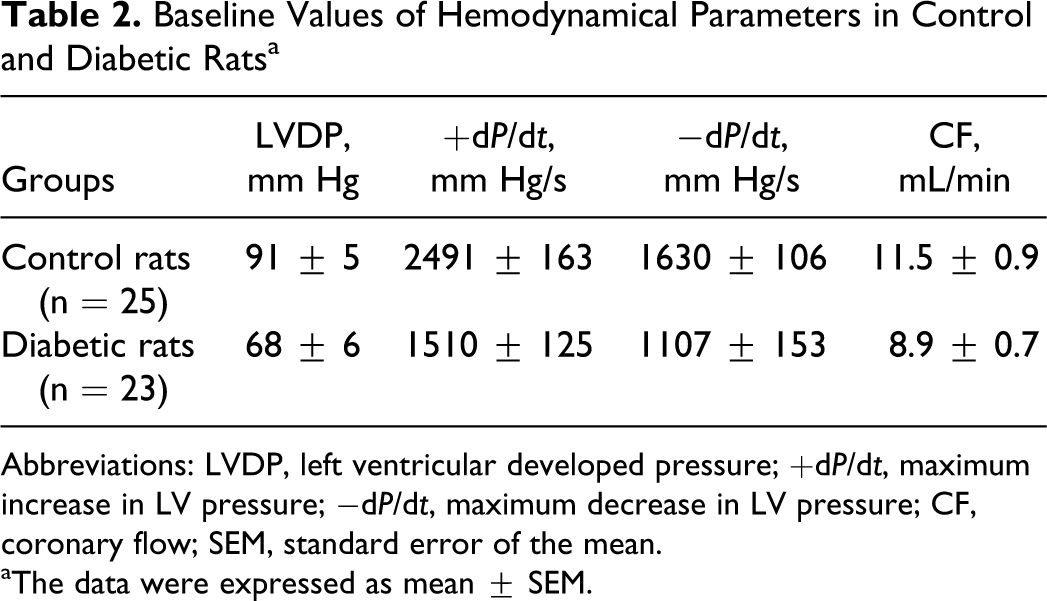
Abbreviations: LVDP, left ventricular developed pressure; +dP/dt, maximum increase in LV pressure; −dP/dt, maximum decrease in LV pressure; CF, coronary flow; SEM, standard error of the mean.
aThe data were expressed as mean ± SEM.
In nondiabetic control animals, IPostC and/or CsA improved LVDP and +dP/dt at some time points during reperfusion (P < .03; Figures 2 and 3). In contrast, either IPostC or CsA failed to restore myocardial function in the diabetic rats. However, administration of CsA in postconditioned diabetic rats significantly returned the cardiac function during the reperfusion toward the preischemic baseline values, similar to that in the nondiabetic control rats (P < .03; Figures 2 and 3). The combination therapy recovered −dP/dt only at 5 minutes of reperfusion in the control and diabetic animals (data not shown). Although IPostC and/or CsA tended to increase the coronary flow in the reperfusion phase, there was no statistically significant difference in the coronary flow between the groups throughout the experiment (coronary flow in mL/min at 5 minutes of reperfusion in control animals: 10.5 ± 0.6 in C + IPostC, 10.8 ± 1.1 in C + CsA, and 9.9 ± 0.9 in C + IPostC + CsA vs 8.8 ± 0.6 in the control group; and in diabetic animals: 7.3 ± 1.2 in D + IPostC, 9.2 ± 1.3 in D + CsA, and 9.1 ± 0.7 in D + IPostC + CsA vs 7.9 ± 0.9 in the diabetic group).

Left ventricular developed pressure (LVDP) alterations in control (top) and diabetic (bottom) animals. The data were expressed as mean ± SEM. #P < .02 difference between C + IPostC and C; †P < .03 difference between C + CsA and C; and *P < .02 difference between C + IPostC + CsA and C (up); *P < .03 difference between D + IPostC + CsA and D (down). C indicates control; IPostC, ischemic postconditioning; CsA, cyclosporine-A; D, diabetic; SEM, standard error of the mean.

Maximum increase in left ventricular pressure (+dP/dt) alterations in control (top) and diabetic (bottom) animals. The data were expressed as mean ± SEM. #P < .03 difference between C + IPostC and C; †P < .03 difference between C + CsA and C; and *P < .02 difference between C + IPostC + CsA and C (up); *P < .03 difference between D + IPostC + CsA and D (down). C indicates control; IPostC, ischemic postconditioning; CsA, cyclosporine-A; D, diabetic; SEM, standard error of the mean.
Infarct Size and RZ
There were no significant differences in the RZ between all groups of control and diabetic animals (Figure 4). Thirty minutes occlusion of the LAD coronary artery produced an IS approximately 41% ± 2.9% of the RZ in control and 39% ± 3.1% in diabetic groups (Figure 5). The differences between IS values of control and diabetic animals were not statistically significant. Both IPostC and CsA significantly reduced the IS of control animals to 28% ± 1.9% and 23% ± 2.0%, respectively (P < .01). However, neither IPostC nor CsA significantly influenced the IS in the diabetic rats (35% ± 1.8% or 32% ± 2.1% vs 39% ± 3.1%, respectively). In nondiabetic animals, IPostC in combination with the administration of CsA shortly prior to the onset of reperfusion had a significant (by 54%) infarct sparing effect (19% ± 1.3% vs 41% ± 2.9%; P < .01). Although IPostC alone could not alter the IS in diabetic rats, combination of CsA and IPostC provided an infarct reducing effect (by 46%) in diabetic groups (21% vs 39%; P < .01; Figure 5).

Risk zone (in percentage of left ventricle) in control (top) and diabetic (bottom) animals. The data were expressed as mean ± SEM, (n = 6). SEM indicates standard error of the mean.

Infarct size (in percentage of risk zone) in control (top) and diabetic (bottom) animals. In nondiabetic animals, ischemic postconditioning alone (C + IPostC), and cyclosporine-A alone (C + CsA) as well as ischemic postconditioning plus cyclosporine-A (C + IPostC + CsA) significantly reduced the infarct size as compared to those of the control (C) group. However, in diabetic animals, only the ischemic postconditioning plus cyclosporine-A (D + IPostC + CsA) significantly reduced the infarct size as compared to those of the diabetic (D) group. The data were expressed as mean ± SEM (n = 6). *P < .01 versus group C (up) or group D (down). C indicates control; D, diabetic; SEM, standard error of the mean.
Nitric Oxide Content
Ischemic postconditioning applied immediately at the onset of reperfusion in the control animals significantly increased myocardial NO levels as compared to nonpostconditioned controls (Figure 6). Administration of CsA in nondiabetic animals increased the NO level nonsignificantly. The production of NO was also significantly enhanced in the group receiving both protective interventions, IPostC and CsA (P < .01). In the diabetic animals, neither IPostC nor CsA influenced the level of myocardial NO production. However, administration of both interventions, IPostC and CsA, significantly enhanced the level of NO in these animals (P < .05; Figure 6).

Nitric oxide metabolites (NOx, μmol/mg protein) level in control (top) and diabetic (bottom) animals. The data were expressed as mean ± SEM. *P < .01 as compared with group C (up) and *P < .05 as compared with group D (down). NOx indicates nitric oxide; C, control; IPostC, ischemic postconditioning; CsA, cyclosporine-A; D, diabetic; SEM, standard error of the mean.
Discussion
In the present study, IPostC and CsA showed protective effects in normal nondiabetic animals by reducing the IS and improving myocardial performance toward baseline values. In 8-week diabetic animals, however, either IPostC or CsA failed to cause the cardioprotection. Nevertheless, with inhibiting the mPTP by means of CsA administration before and in the early periods of the reperfusion, the cardioprotective effect of IPostC was achieved in the diabetic animals, and this response was comparable to those of the normal animals. In addition, administration of IPostC and/or CsA could significantly increase NO levels of the nondiabetic myocardium. In contrast, only the combination of IPostC and CsA revealed a sparing effect on IS and enhanced myocardial NO production in diabetic hearts.
The majority of studies carried out on the cardioprotective effect of IPostC have been performed under normal conditions on healthy animals without any risk factors. 5,19 –21 Clinically, most of the patients with ischemic heart disease, however, also have comorbidities, including diabetes, which may interact with ischemic injury. 7 Therefore, investigating the protective effect of IPostC strategies in the presence of cardiovascular risk factors is of particular interest and can clinically provide much more reliable results applicable to humans. Diabetes mellitus is one of the most frequent risk factors for MI, and diabetic patients are more prone to cardiac ischemic dysfunction, including I/R injury. 1,2 Interaction of the diabetic state with therapeutic strategy of IPostC has not been studied in basic and clinical circumstances.
The body of literature has documented that inhibition of mPTP acts as a pivotal end effector for many of the protective ischemic and pharmacological preconditioning and postconditioning strategies. 8,9,11 The mPTP opens during the onset of reperfusion and leads to disruption of mitochondrial integrity resulting in worsening of I/R injury. 20 In the present experiment, while all therapeutic strategies were protective in healthy animals, IPostC or CsA alone failed to provide any protection in diabetic hearts. However, only combination of both CsA and IPostC showed full cardioprotection in diabetic rats. These findings indicate that IPostC may exert its cardioprotective effect partly by inhibiting mPTP in normal animals, but it seems that the effectiveness of IPostC in the diabetic rats is not as strong as in the normal state to activate endogenous prosurvival signal transduction mediators in an effective way to cause cardioprotection. After bringing CsA into play, however, the effects of IPostC and CsA appear additive, and their final cardioprotective effects are enhanced. Most importantly, this finding raises the possibility of an involvement of protective pathways other than mPTP inhibition by postconditioning as well, which are not examined in this study. Postconditioning can activate different subcellular mediators, including mitogen activated protein kinase-extracellular regulated kinase 1/2 (MEK-ERK1/2), protein kinase C (PKC), signal-transducer and activator of transcription-3 (STAT3), and endothelial NO synthase (eNOS) which are not necessarily terminating on mPTP, rather they can also activate protein translation or lead to the opening of mitochondrial ATP-dependent potassium (mKATP) channel and cause cardioprotection. 21 –24 Furthermore, postconditioning may strengthen the inhibition of the mPTP by CsA. Finally, to determine the exact reason for these additional effects, further studies are warranted.
The exact mechanisms responsible for the failure of IPostC to produce cardioprotection in diabetic rats are not understood. Nitric oxide plays multiple roles in the cardiovascular system, mediating a number of physiological and pathophysiological processes. Nitric oxide is one of the endogenous mediators which contribute to myocardial protection in ischemic preconditioning and postconditioning. 12 In the present study, IPostC failed to increase the amount of NO in diabetic rats, and this failure can, to some extent, explain the loss of cardioprotection in these animals. The increased amount of NO production by combining IPostC and CsA was associated with a cardioprotective effect in both diabetic and nondiabetic animals. The endogenous production of NO by postconditioning in the setting of I/R directly or indirectly can somewhat reduce the reperfusion-induced cardiac dysfunction, presumably by reducing the diastolic stiffness, inflammatory mediators, and oxygen consumption, 25,26 activating the protein kinases such as PKG and PKC, 27,28 and inhibiting apoptosis by acting on mitochondria. 28,29 Nitric oxide may also have other positive actions by dilating the coronary vessels and increasing coronary flow, which in turn improves the circulation of the perfusion solution in different parts of the heart, thus preventing the fall in LVDP during reperfusion.
Nitric oxide exhibits a cross-link with protective signaling pathways, including PI3K/Akt pathway to inhibit mPTP and provide tissue protection. 28 Activation of PI3K/Akt and ERK pathways by IPostC leads to phosphorylation of NOS and enhanced production of NO. It is reported that increased level of NO is associated with the opening of mKATP channel, which is also cardioprotective. 30 Diabetes mellitus might negatively affect these crucial pathways of postconditioning. In agreement with this hypothesis, ERK and GSK-3β located upstream in the signal transduction pathway of the mPTP and mKATP channel were not phosphorylated in diabetic animals compared to the healthy controls. 31 Increasing the potency of postconditioning strategy, for example, concomitant application of 2 protective interventions, IPostC and CsA, can overcome the diabetes-induced alterations in signaling pathways and result in cardioprotection. Moreover, abnormal production of factors, including free radicals, inflammatory mediators, and myocardial dysfunction in diabetes mellitus 32 or other neurohumoral alterations, which could not be investigated in the isolated heart setting can act as interfering elements and block the effective cardioprotection by IPostC. Further research is needed to clarify the share of these mechanisms contributing to the failure of cardioprotective effect of IPostC in diabetes.
In conclusion, the findings of the present study indicate that IPostC with 3 cycles of 30 seconds R/I at the onset of reperfusion or administration of CsA fails to increase NO production and fails to protect the diabetic myocardium against I/R injury in rats. This loss of cardioprotection can be recovered by the concomitant administration of CsA and IPostC at reperfusion.
The author(s) declared no conflicts of interest with respect to the research, authorship, and/or publication of this article.
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was partly supported by Drug Applied Research Center, Faculty of Medicine, Tabriz University of Medical Sciences, Tabriz, Iran.