
Abstract
Ketamine is a unique anesthetic agent that induces dissociative anesthesia, characterized by perceptual detachment, analgesia, and altered states of consciousness. Beyond its widespread use in anesthesia, subhypnotic ketamine dosing has emerged as a rapid-acting antidepressant and a valuable model for probing the neural mechanisms underlying consciousness and neuropsychiatric disorders. At the core of its effects are actions on cortical circuits, primarily through NMDA receptor and HCN1 channel antagonism, disinhibition of pyramidal neurons, and altered thalamocortical connectivity. This review brings together emerging findings from ketamine pharmacology, cell type–resolved and region-specific in vivo imaging, and systems neuroscience to define how ketamine alters cortical circuit dynamics to drive dissociation. We further explore the intriguing possibility that ketamine freely diffuses into and concentrates within intracellular compartments and, in doing so, modulates neuronal excitability, intracellular signaling, and an epigenetic state, even following a single dose. A deeper mechanistic understanding of these cortical and cellular processes will not only advance our knowledge of ketamine’s complex pharmacology but may also inform new therapeutic strategies for treatment-resistant depression and facilitate the study of diverse states of consciousness.
Introduction
Racemic (R,S)-ketamine was first introduced in the 1960s as a safer alternative to phencyclidine (PCP), offering anesthesia with a more favorable safety profile (Domino et al 1965). Over subhypnotic to anesthetic doses, ketamine produces a unique state of consciousness characterized by sensory detachment, depersonalization, and distorted perception of space and time—collectively termed dissociation (Domino et al 1965; Krystal et al 1994). At anesthetic doses, ketamine induces a dense state of unresponsiveness compatible with surgery in animals and humans. Unlike conventional anesthetics that produce unconsciousness, however, ketamine anesthesia is associated with vivid sensory experiences completely divorced from reality. This peculiar ketamine-induced state is the reason why ketamine is classified as a “dissociative anesthetic” (Corssen and Domino 1966; Domino 1980; Olofsen et al 2022; Dávila et al 2025). Ketamine’s psychoactive effects, while initially considered undesirable, are now recognized as clinically and mechanistically significant, perhaps contributing to its clinical utility. In recent years, subanesthetic ketamine has demonstrated rapid and sustained antidepressant effects, particularly in treatment-resistant depression (Berman et al 2000; Zarate et al 2006; Price et al 2009; Murrough et al 2013; Fava et al 2020). Some studies suggest that dissociation could be a “biomarker” for having triggered sufficient cortical modulation to mediate rapid antidepressant effects (Luckenbaugh et al 2014; Ballard and Zarate 2020). At anesthetic doses, the dissociative and analgesic effects of ketamine appear to be closely linked (Olofsen et al 2022). These therapeutic properties have reinvigorated efforts to understand the neurobiological basis of ketamine’s dissociative actions, particularly within cortical circuits.
Cortical mechanisms are central to the dissociative state (Cichon et al 2023). By blocking open NMDA receptors and HCN1 channels, ketamine perturbs the excitatory–inhibitory balance of the cortex, most notably by preferentially inhibiting NMDA receptors on fast-spiking GABAergic interneurons (Homayoun and Moghaddam 2007; Gerhard et al 2020; Hu et al 2025). This disinhibition leads to a cascade of downstream effects, including increased pyramidal neuron excitability, desynchronized oscillatory activity, and disrupted thalamocortical communication (Vesuna et al 2020). Recent studies have demonstrated that ketamine differentially modulates neuronal activity across cortical layers and even brain regions, supporting the existence of a finely tuned, layer- and region-specific mechanism of action (Vesuna et al 2020; Hu et al 2025). However, this specificity may diminish at higher doses, where the effects appear to become more uniform across cortical circuits (Cichon et al 2023).
Dissociation offers a powerful window into the neural basis of consciousness, and ketamine serves as a pharmacologic tool and a therapeutic agent. Yet, understanding how the many and diverse effects of ketamine on distinct neuronal subtypes and circuits combine to give rise to the dissociated state is presently incomplete. Here, we synthesize current knowledge on ketamine pharmacology and the cellular and circuit-level mechanisms that generate ketamine-induced dissociation, focusing specifically on the cortical dynamics that support this altered state. We integrate classical findings with emerging work from in vivo brain circuit imaging, electrophysiology, and molecular approaches—including recent studies identifying layer- and cell type–specific changes in response to ketamine administration.
Molecular Targets of Ketamine in Cortical Neurons
Ketamine’s primary pharmacologic action is antagonism of the NMDA receptor (binding at the same site as PCP, deep within the ion channel pore), a subtype of glutamate receptor that is critical for synaptic plasticity and excitatory neurotransmission (Zorumski et al 2016; Y. Zhang et al 2021). This open (ie, activity dependent) channel blockade is noncompetitive and voltage dependent, leading to reduced synaptic excitation in vitro (Anis et al 1983; MacDonald et al 1987; Orser et al 1997). NMDA receptors are expressed across excitatory and inhibitory neurons, but ketamine appears to preferentially block NMDA receptors on fast-spiking parvalbumin (PV)– and somatostatin (SST)–expressing interneurons due to their high baseline activity and tonic depolarization (Homayoun and Moghaddam 2007; Seamans 2008; Ali et al 2020; Gerhard et al 2020). This selectivity disrupts local inhibitory control within cortical microcircuits, particularly in the prefrontal cortex, resulting in enhanced activity of downstream pyramidal neurons (Li et al 2010). Additionally, NMDA receptor antagonism suppresses long-term potentiation and alters the balance of synaptic scaling (Duman and Aghajanian 2012). While evidence suggests that its antidepressant and dissociative anesthetic properties are mediated by NMDA receptor antagonism (Autry et al 2011; Duman et al 2012; Kadriu et al 2021), the fact that other NMDA antagonists do not elicit the same effects indicates that activity at additional receptors may also contribute (Zorumski et al 2016).
An additional critical contributor to ketamine’s cortical actions is its effect on hyperpolarization-activated cyclic nucleotide–gated (HCN) channels, which regulate neuronal excitability and rhythmic activity (Magee 1998; Chen et al 2009; Wahl-Schott and Biel 2009; Duman and Aghajanian 2012). HCN1 channels are expressed widely across the cortex and are particularly enriched in layer 5 pyramidal neurons and inhibitory interneurons (Lörincz et al 2002). Ketamine has been shown to inhibit HCN1 channel currents, reducing the hyperpolarization-activated inward current (Ih) that stabilizes resting membrane potential and shapes dendritic integration (Chen et al 2009). This inhibition can lead to enhanced excitability of excitatory and inhibitory cells, depending on their intrinsic properties and network context (Lewis et al 2024). In pyramidal neurons, reduced HCN1 conductance increases dendritic input summation and may facilitate bursting behavior, while in interneurons, it may exacerbate disinhibition (Santoro et al 2000). The suppression of HCN1 channels by ketamine may thus amplify or sustain cortical destabilization initiated by NMDA receptor blockade, contributing to the overall emergence of the dissociative state (Vesuna et al 2020; Cichon et al 2023; Hu et al 2025).
In addition to the well-studied effects on the NMDA receptors and HCN channels, ketamine binds to sigma receptors and opioid receptors, as well as serotonin and norepinephrine transporters, although the functional relevance of these interactions remains unclear (Sleigh et al 2014). Some studies suggest that its antidepressant effects may involve a combination of NMDA receptor blockade and secondary signaling cascades, including BDNF release and mTOR activation triggered by persistent neuronal activity (Li et al 2010). These noncanonical mechanisms could contribute to the delayed but lasting synaptic remodeling observed following a single dose of ketamine (Phoumthipphavong et al 2016).
Ketamine and PCP Metabolism and Dissociative Actions
Ketamine was first synthesized in 1962 by chemist Calvin Stevens at the University of Michigan as part of an effort to develop a safer alternative to PCP, or “angel dust” (Kusljic et al 2016; Hashimoto 2022). Ketamine is rapidly metabolized (t1/2 = 45 min) in the liver via CYP2B6 and CYP3A4 into norketamine (NK), hydroxynorketamine (HNK), and dehydronorketamine (DHNK; Figures 1 and 2). While NK retains NMDA antagonism, HNK has minimal NMDA blocking activity and exerts antidepressant effects via the TrkB receptor (Casarotto et al 2021) and mTOR pathway activation (Paul et al 2014), suggesting that ketamine’s antidepressant actions may be separable from its dissociative properties. (2R,6R)-HNK lacks the sensory dissociative effects and abuse potential that limit the broader therapeutic use of ketamine (Figure 1; Gould et al 2019). (2S,6S)-HNK may also retain the antidepressant properties (Figure 2; Moaddel et al 2013; Kawatake-Kuno et al 2024). Unlike ketamine or NK, DHNK has negligible affinity for NMDA receptors (Ki = 38.95 μM for [S]-DHNK; Moaddel et al 2013) and does not contribute to ketamine’s dissociative effects, which are primarily mediated by NMDA receptor antagonism Although initially regarded as inactive at the receptor, DHNK has since been identified as a potent and selective negative allosteric modulator of the α7-nicotinic acetylcholine receptor (half maximal inhibitory concentration, 55 nM; Kusljic et al 2016; Guhathakurta et al 2024). Preclinical studies indicate that DHNK lacks central nervous system activity associated with dissociation or psychotomimetic side effects and does not trigger antidepressant activity in animal models (Sałat et al 2015; Zanos et al 2018).
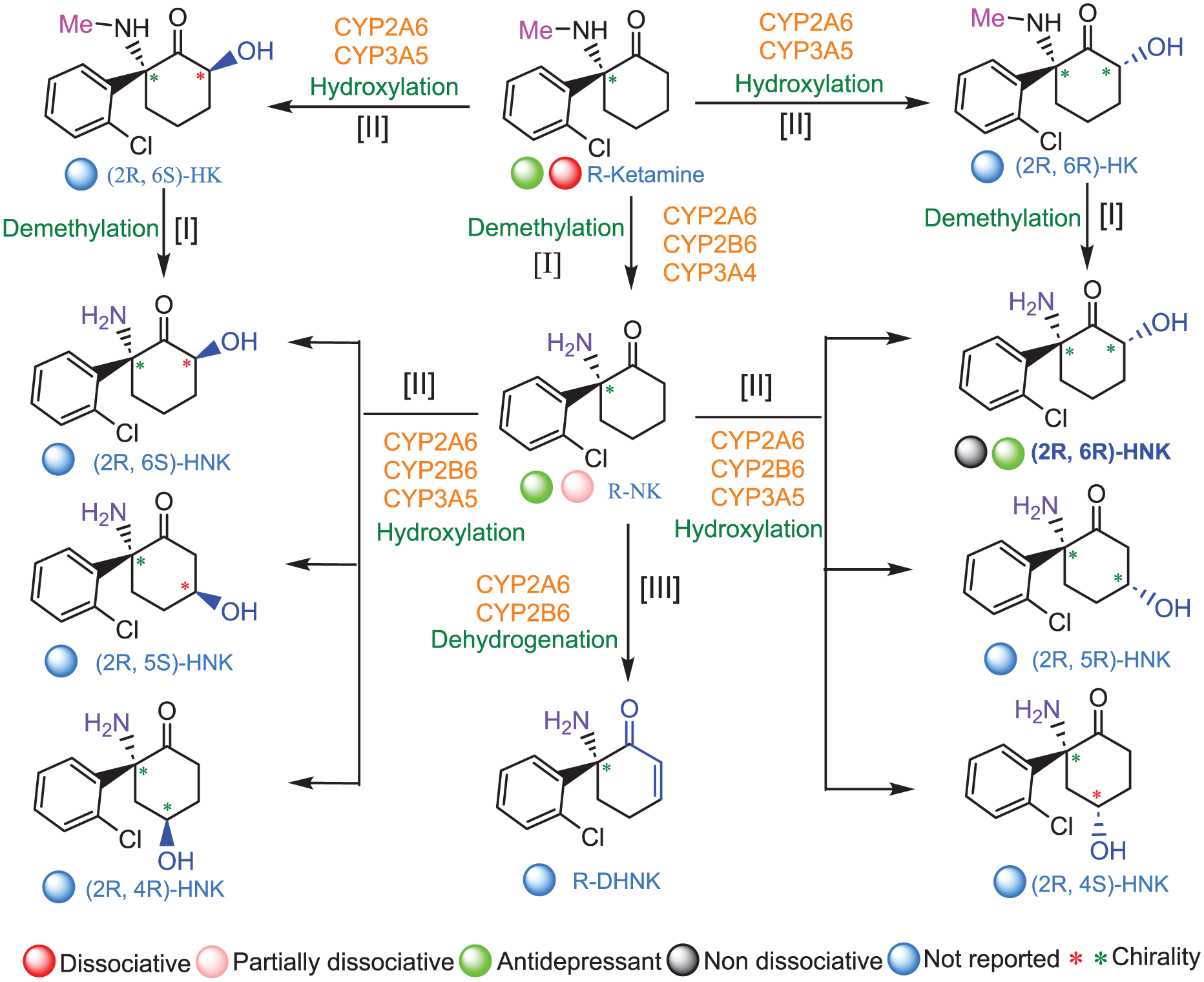
In vivo metabolism of R-ketamine. R-ketamine undergoes a series of intricate metabolic transformations in vivo, primarily orchestrated by cytochrome P450 enzymes. The first major step is demethylation (step I), which converts the parent compound into (R)-norketamine (NK). In parallel, R-ketamine can be hydroxylated (step II) at the C6 position of the cyclohexanone ring, generating 2 classes of stereoisomeric 6-hydroxyketamines (HK): (2R,6R)/(2R,6S)-HK. From there, the metabolic landscape further diversifies. (R)-norketamine can undergo dehydrogenation (step III), yielding dehydronorketamine (DHNK), or be further hydroxylated to produce a rich variety of hydroxynorketamine (HNK) isomers. These include stereoisomers such as (2R,6R)/(2R,6S)-HNK, as well as additional variants including (2R,5R)/(2R,5S)- and (2R,4R)/(2R,4S)-HNK. The hydroxylated ketamine derivatives themselves—(2R,6R)- and (2R,6S)-HK—can also undergo demethylation, forming their corresponding HNKs. The intriguing chemical structure of R-ketamine that involves metabolic branching contributes to its complex pharmacologic actions, with each metabolite potentially carrying distinct functional properties (Zanos et al 2016, 2018).
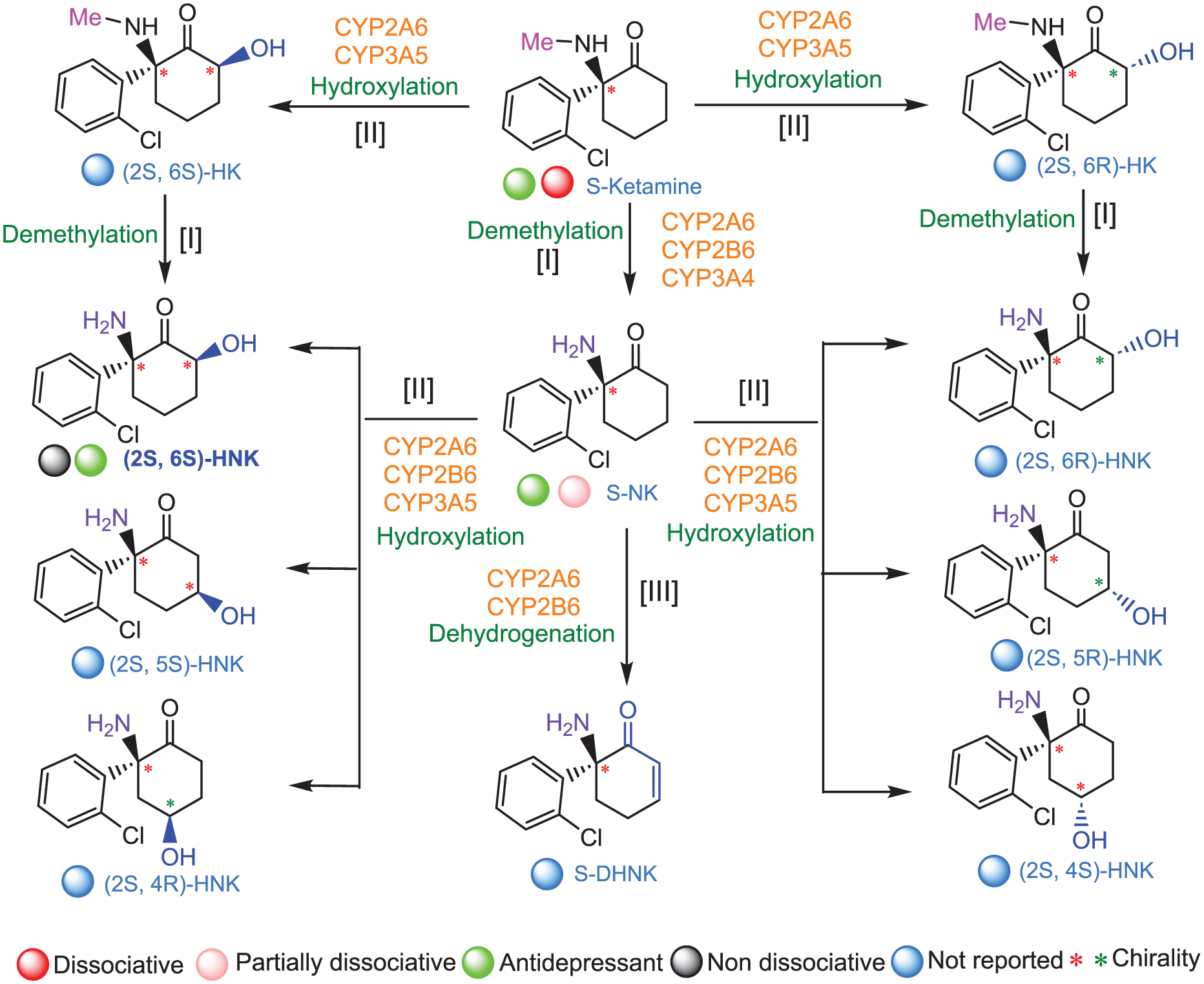
In vivo metabolism of S-ketamine. S-ketamine undergoes a series of intricate metabolic transformations in vivo, primarily orchestrated by cytochrome P450 enzymes. The first major step is demethylation (step I), which converts the parent compound into (S)-norketamine (NK). In parallel, ketamine can be hydroxylated (step II) at the C6 position of the cyclohexanone ring, generating 2 classes of stereoisomeric 6-hydroxyketamines (HK): (2S,6R)- and (2S,6S)-HK. From there, the metabolic landscape further diversifies. (S)-Norketamine can undergo dehydrogenation (step III), yielding dehydronorketamine (DHNK), or be further hydroxylated to produce a rich variety of hydroxynorketamine (HNK) isomers. These include stereoisomers such as (2S,6R)/(2S,6S)-HNK, as well as additional variants including (2S,5R)/(2S,5S)- and (2S,4R)/(2S,4S)-HNK. The hydroxylated ketamine derivatives themselves—(2S,6R)- and (2S,6S)-HK—can also undergo demethylation, forming their corresponding HNKs. The intriguing chemical structure of S-ketamine that involves metabolic branching contributes to its complex pharmacologic actions, with each metabolite potentially carrying distinct functional properties (Zanos et al 2016, 2018).
PCP has a much longer half-life (t1/2 = 12–24 h) and generates poorly characterized metabolites that likely do not contribute to therapeutic action, resulting in prolonged dissociation and higher abuse potential (Kapur and Seeman 2002; Rocha et al 2017; Bates and Trujillo 2021). Both drugs increase dopaminergic activity in the mesolimbic system, but PCP has stronger dopamine transporter inhibition, accounting for its psychotomimetic and behavioral toxicity (Moaddel et al 2013). Electroencephalogram recordings demonstrate that ketamine and PCP, as NMDA receptor antagonists, induce characteristic increases in gamma-band activity and reductions in alpha oscillations (Sebban et al 2002; Akeju et al 2016; Amat-Foraster et al 2019; Adam et al 2024). These frequency changes likely reflect cortical network disinhibition common to NMDA receptor hypofunction. Clinically, the dissociative effects of ketamine are transient and dose dependent, making it suitable for controlled therapeutic use in anesthesia and depression treatment applications, whereas PCP’s severe, unpredictable dissociation precluded its medical use. However, PCP remains a preclinical model for studying NMDA receptor hypofunction and schizophrenia (Kapur and Seeman 2002; Bertron et al 2018). Understanding the distinct metabolic and neurocircuit mechanisms of ketamine and PCP is essential for advancing dissociative neuroscience and developing next-generation treatments that promote therapeutic benefits while minimizing severe dissociative side effects.
Ketamine with plasma concentration 500 to 1000 ng/mL induces nearly all of the dissociative and psychotomimetic effects (Figure 3A) associated with NMDA receptor antagonism (Ki = 324 nM, S-ket; 1.4 µM, R-ket), whereas its primary metabolite, NK (Ki = 1.7 µM, S-NK; 13 µM, R-NK), elicits only 5% to 10% of these effects in humans (Zanos et al 2018). Despite this divergence in neuropsychiatric outcomes, NK is structurally similar to ketamine, differing only by the removal of a methyl group at the C-2 position on the cyclohexanone ring, where methylamine is replaced by a primary amine. The demethylation of ketamine to NK is rapid, yet the fate of the liberated methyl (CH3+) group at dissociative doses remains unknown. It is plausible that ketamine functions as a methyl donor in some capacity or that the released methyl group is metabolized to methanol and subsequently oxidized to formaldehyde (Wang et al 2019). As formaldehyde is capable of crosslinking or further methylating proteins, DNA, and RNA (Lu et al 2008; Edrissi et al 2013; Chen et al 2017; Feng et al 2025), this pathway could represent an additional, non–receptor-mediated mechanism contributing to ketamine’s dissociative effects and neurotoxicity. Several research groups are now investigating additional, previously uncharacterized ketamine metabolites (Vallianatou et al 2024) that may also contribute to dissociation—a logical direction, given the inherent instability of HNK derivatives and their propensity to oxidize to diketone species (Muschallik et al 2020; Vallianatou et al 2024).
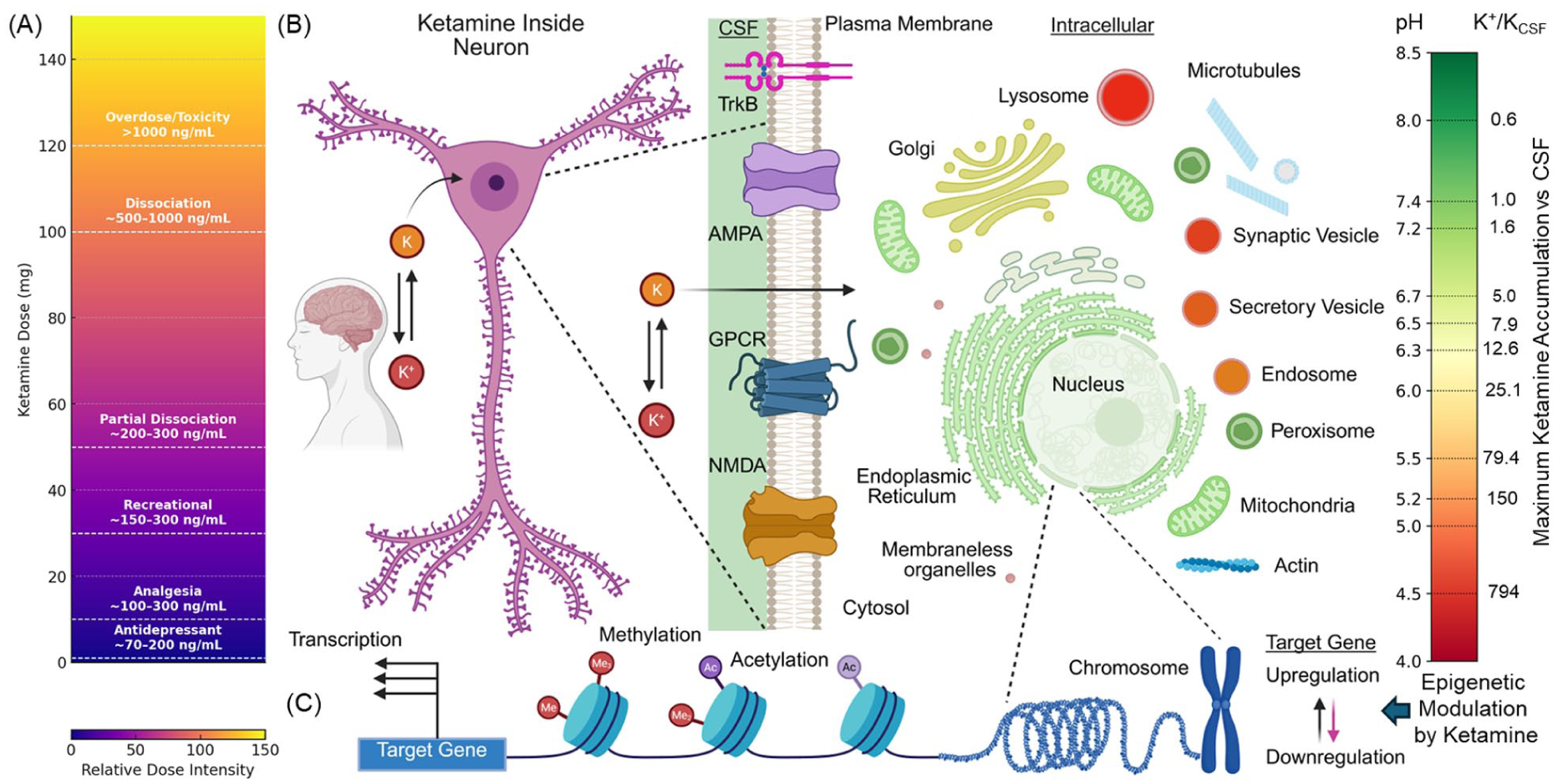
(A) Ketamine dosage vs effect continuum with plasma concentration for a 70-kg adult. A gradient visualization illustrates the progressive effects of ketamine as dosage increases. The color gradient reflects rising dose intensity from bottom to top or left to right. Vertical dashed lines mark key transition points between subjective and physiologic effect zones. At 0.5 to 1 mg, Cmax plasma = 70 to 200 ng/mL, and ketamine produces antidepressant properties. At 10 mg, Cmax plasma = 100 to 300 ng/mL, and ketamine produces mild analgesia. At 30 mg, Cmax plasma = 150 to 300 ng/mL, and users experience recreational perceptual changes. At 50 mg, Cmax plasma = 200 to 300 ng/mL, and partial dissociation begins. At 100 mg, Cmax plasma = 500 to 1000 ng/mL, which leads to full dissociation (often referred to as the “K-hole”). Doses beyond 100 mg (Cmax plasma >500-1000 ng/mL) may result in profound dissociation, anesthesia, or neurotoxic risk. This continuum emphasizes the narrow therapeutic window between clinical and potentially harmful effects. (B) Ketamine (K) inside neurons and its plausible subcellular distribution. Ketamine quickly crosses neuronal membranes in its neutral form and accumulates in acidic intracellular compartments through acid trapping of its protonated form (K+). The heat map shows predicted subcellular ketamine accumulation (K+/KCSF) based on organelle pH, highlighting a shift from traditional surface receptor targeting to intracellular mechanisms of action, as predicted by de Duve et al (1974). The vesicles are not drawn to scale (Blumenfeld et al 2024). (C) Epigenetic cascade. Ketamine’s subsequent intracellular signaling leads to increased histone acetylation (Ac)–deacetylation and DNA methylation–demethylation at promoters of neuroplasticity-related genes. Ketamine also alters microRNA expression, regulating posttranscriptional control of proteins involved in synaptic structure and function. These epigenetic changes create a permissive chromatin state, enhancing the transcription of genes critical for synaptic remodeling. The resulting long-term changes include dendritic spine growth, enhanced synaptogenesis, and circuit reconfiguration, contributing to ketamine’s rapid antidepressant effects and, at higher doses, dissociative state. This figure was made with BioRender.
Rapid Intracellular Detection and Accumulation of Ketamine
The weakly basic (pKa = 7.5), deprotonated form of ketamine readily enters neurons due to its small size and lipophilicity (log P = 2.2) at physiologic pH. The deprotonated form of ketamine readily diffuses across the plasma membrane, enabling passive intracellular entry. Once internalized, ketamine accumulates within acidic organelles (lysosomes, synaptic vesicles, and endosomes) and, to a lesser extent, in the endoplasmic reticulum (ER) via a mechanism consistent with ion trapping (Lester et al 2015; Villéga et al 2024). In these compartments, the amine group becomes reprotonated, preventing the molecule from diffusing back out and effectively trapping it intracellularly—a concept first proposed by De Duve to explain the accumulation of weak bases within subcellular organelles (Blumenfeld et al 2024). The theoretical maximal accumulation of a weakly basic drug with a single protonation site, versus blood, increases by 10-fold for every unit of difference between the pH of the organellar lumen and that of the blood (Figure 3B).
In support of this hypothesis, Bera et al (2019) designed and optimized genetically encoded fluorescent ketamine biosensors (iSKetSnFR1 and iSKetSnFR2) to measure the subcellular pharmacokinetics of S-ketamine. Plasma membrane– and ER-targeted sensors revealed that S-ketamine rapidly enters and equilibrates within the ER—within seconds—in Neuro2a cells and human induced pluripotent stem cell–derived dopaminergic neurons. ER concentrations closely tracked extracellular levels but accumulated only modestly, consistent with the reduced proton-trapping effect in the relatively neutral ER environment (Figure 3B). These observations implicate intracellular compartments, including organelles, as plausible sites of S-ketamine target engagement, potentially involving organellar ion channels, receptors, or transporters, and suggest a possible role for these interactions in mediating its antidepressant, anesthetic, and dissociative effects (Lester et al 2015).
An adequate theory of acid trapping must consider the efficiency of proton pumps that maintain organellar pH, the compound’s pKa, the low membrane permeability of its charged form, the charge state of the protonated drug, the organellar membrane potential, and additional contributing factors (de Duve et al 1974). At dissociative doses, ketamine may perturb the biophysical properties of neuronal membranes by altering lipid fluidity, thereby influencing the conformation, distribution, and activity of membrane-associated receptors and ion channels (Kapoor et al 2019). Such effects on synaptic transmission, neuronal excitability, and intracellular signaling pathways could underlie the emergence of dissociative phenomena at higher doses (Blumenfeld et al 2024). At dissociative concentrations, the weakly basic nature of ketamine may also disrupt the proton gradient within acidic organelles, raising their intraluminal pH and potentially disrupting the folding and function of ion channels, receptors, and other intracellular proteins (Ohkuma and Poole 1978). Such alterations could underlie the molecular basis of ketamine’s dissociative effects. Testing this hypothesis will require the development of next-generation acid-tolerant chemical and genetically encoded biosensor scaffolds.
Ketamine’s intracellular retention may have direct consequences for cortical circuitry. Endosomal and lysosomal compartments shape the trafficking and turnover of AMPA and NMDA receptors, and ion trapping–mediated accumulation of ketamine within these acidic organelles could shift these pathways toward enhanced AMPA receptor surface insertion or altered NMDA receptor subunit routing. These receptor-level changes would be expected to increase excitatory synaptic gain and cortical excitability, providing a mechanistic link among intracellular ketamine dynamics, dissociation-related circuit activation, and the synaptic plasticity thought to underlie its antidepressant efficacy.
Ketamine-Induced Changes in Cortical Microcircuits
At the cellular level, subhypnotic, antidepressant doses of ketamine acutely enhance glutamatergic transmission (Moghaddam et al 1997; Abdallah, De Feyter, et al 2018), augment postsynaptic responses (Ali et al 2020), and increase the firing rates of cortical pyramidal neurons (Figure 4, top panel; Patel and Chapin 1990; Homayoun and Moghaddam 2007; B. Zhang et al 2021). Over longer timescales, these doses promote synaptic plasticity across multiple brain regions (Moda-Sava et al 2019). In parallel, ketamine alters activity within a complex and highly interconnected network of cortical GABAergic interneurons, which may contribute critically to its effects (Figure 4, top panel). Notably, fast-spiking PV- and SST-expressing interneurons are rapidly inhibited following ketamine administration or exposure to selective NMDA receptor antagonists (Figure 3). The suppression of PV interneurons, which primarily target the perisomatic region of pyramidal cells, and SST interneurons, which inhibit distal dendrites, may lead to a net disinhibition of pyramidal neurons, thereby facilitating ketamine’s proexcitatory and circuit-modifying actions (Homayoun and Moghaddam 2007; Ng et al 2018; Ali et al 2020; Gerhard et al 2020; B. Zhang et al 2021; Qiao et al 2024).
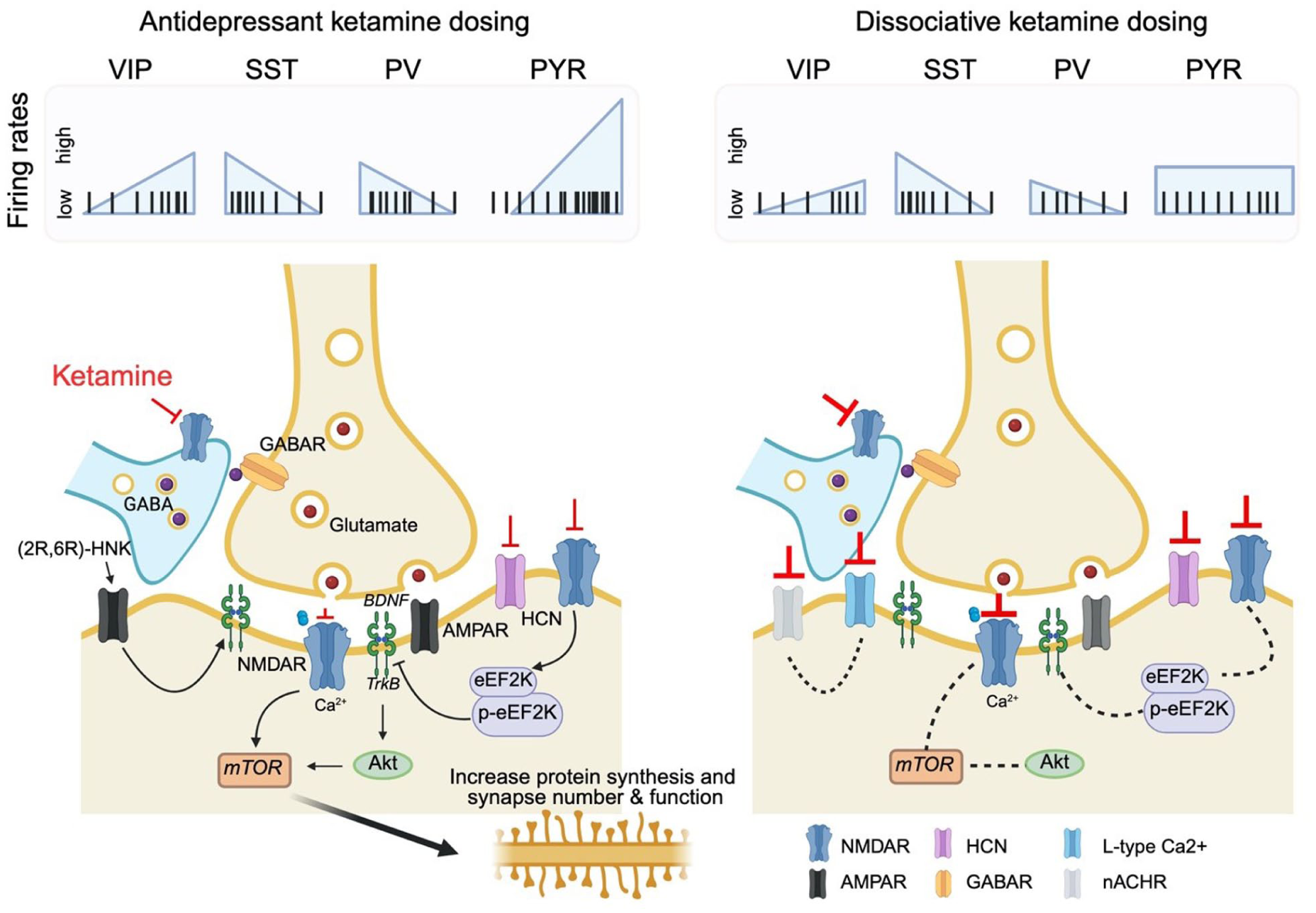
Ketamine-induced cellular and molecular effects in cortical microcircuits. Top: Simplified schematic of ketamine-induced changes in the neuronal activity of major cortical cell types, including GABAergic interneuron subtypes—vasoactive intestinal peptide (VIP+), somatostatin (SST+), and parvalbumin (PV+) neurons—as well as excitatory pyramidal neurons, at antidepressant vs dissociative doses. VIP+ interneurons disinhibit pyramidal cells by suppressing SST+ neurons, which normally inhibit pyramidal dendrites. PV+ interneurons target the perisomatic region of pyramidal neurons and regulate their output. Bottom: At antidepressant doses (left), ketamine (red inhibitor) engages a disinhibitory microcircuit that enhances glutamate release and pyramidal neuron depolarization, driving calcium entry and activation of molecular cascades that increase protein synthesis, synapse number, and synaptic function. At dissociative doses (right; the thicker inhibitor line indicates higher ketamine dose relative to the antidepressant dose), ketamine produces similar interneuron modulation, but overall pyramidal activity remains stable due to a redistribution of activity: previously silent neurons become active while formerly active neurons fall silent (Cichon et al 2023). The molecular signaling underlying dissociative dosing remains poorly defined in preclinical models (dashed lines). Figure created with BioRender.
In contrast to the well-characterized molecular and circuit changes induced by antidepressant doses of ketamine, the effects of dissociative doses have only recently begun to be described (Figure 4, bottom panel). Using high-speed, brain-wide recording approaches, Vesuna et al (2020) revealed dissociative doses of ketamine-induced layer- and region-specific changes in cortical activity. Ketamine and PCP elicited a 1- to 3-Hz traveling wave of activity initiated in layer 5 neurons of the retrosplenial cortex (RSC). Rhythmic optogenetic activation of RSC layer 5 neurons recapitulated dissociation-like behavioral effects. Interestingly, local RSC HCN1 activity was required for ketamine to induce the slow oscillation and dissociation-like behavioral effects. Simultaneous recordings from high-density probes showed that the RSC remained rhythmically coupled to connected thalamic circuits while becoming uncoupled from most other brain regions, including an inverse correlation with frontally projecting thalamic nuclei. Strikingly, Vesuna et al reported comparable oscillatory activity in a patient who spontaneously exhibited dissociative symptoms during the prodromal phase of a seizure. This foundational work identifies the RSC as a key locus for dissociation and shows that HCN1 is required for ketamine to induce slow rhythms and dissociation-like behaviors.
In a recent study, Hu et al (2025) showed that dissociative doses of ketamine selectively inhibit PV interneurons in the RSC, producing slow electroencephalogram (delta band) oscillations and dissociation-like behaviors in mice. Optogenetic suppression of RSC PV interneurons was sufficient to evoke ketamine-like oscillatory activity and partially recapitulate dissociation-related behaviors in mice. Conversely, activating RSC PV interneurons, or knocking down the NMDA receptor subunit NR1 or HCN1 channels in these cells, attenuated ketamine-induced delta oscillations and behavioral readouts. With Vesuna et al (2020), these findings implicate RSC slow oscillations as a requisite network feature of ketamine dissociation and suggest that layer 5 pyramidal neurons and PV interneurons contribute via NMDA receptor– and HCN1-dependent mechanisms. The precise circuit interactions between these cell types during dissociation, however, remain to be defined.
Work from Cichon et al (2023) demonstrated that dissociative doses of ketamine induce a striking reconfiguration of excitatory neuronal activity across the neocortex, characterized by a switch in which spontaneously active neuronal ensembles are silenced while previously quiescent neurons become active (Figure 5A; Mathew and Zarate 2016; Abdallah, Sanacora, et al 2018). The net neuronal activity of pyramidal neurons at dissociative ketamine doses seems to be no different than during wakefulness (Schroeder et al 2016; Cichon et al 2023). This shift occurs across all cortical layers and regions examined and is mediated by suppression of PV+ and SST+ interneuron activity, along with inhibition of NMDA receptor and HCN1 channel function. Building on these findings, a working model has been proposed in which this neuronal switch emerges from 2 interrelated phenomena: the suppression of previously active NMDAR-dependent neurons and the activation of formerly silent pyramidal neurons (Figure 5B). The latter are disinhibited through ketamine-sensitive GABAergic interneurons and exhibit enhanced AMPAR-mediated synaptic responses due to HCN1 channel inactivation. This dual mechanism impairs dendritic integration in active neurons while potentiating activity in typically dormant neurons, thus fragmenting normal circuit motifs and promoting novel, complex patterns of activity that are largely independent of thalamic input. This cortical reorganization likely underlies the dissociative phenomenology associated with ketamine by disrupting integration within and across layers and shifting the cortex toward an abnormal brain state.
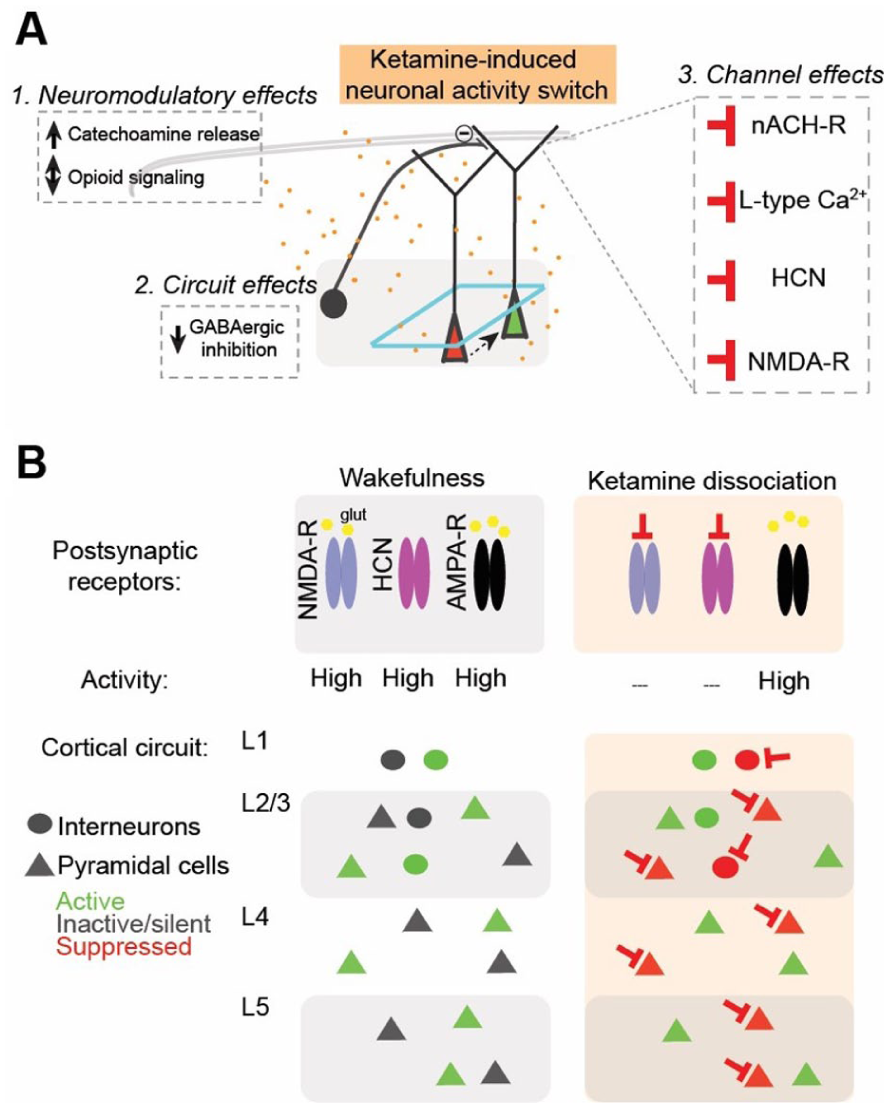
Neocortical mechanisms contributing to dissociation. (A) Molecular, cellular, and circuit changes in the neocortex underlying ketamine dissociation. Ketamine simultaneously alters key ion channels regulating neuronal excitability, modulates inhibitory interneuron function, and reshapes the spontaneously active populations of pyramidal neurons (red cells switching to green). (B) A working model for how ketamine reshapes the spontaneously active populations of pyramidal neurons in cortical microcircuits to contribute to the disconnected state of dissociation. In wakefulness (left), a network of active cells (green) is mostly driven by glutamatergic receptor activation, AMPAR and NMDAR, and modulated by HCN1 activity. Under ketamine (right), NMDAR conductances are inhibited (red inhibitory arrows), transitioning previously active cells into a low-firing mode (green cells turned red). Neighboring neurons, in the setting of reduced GABAergic inhibitory tone and HCN inhibition, efficiently summate a new set of ketamine-driven inputs/synaptic responses, yielding activation (new set of green cells). The shift away from an NMDAR-predominant neurotransmission to AMPAR and a change in inputs drive the neuronal activity of a different network of neurons, creating a new unique pattern of ketamine-dependent circuit activity across the cortical mantle, contributing to ketamine’s disconnected state. Reproduced with permission from Cichon et al (2022), Nature Neuroscience.
Large-Scale Brain Dynamics Induced by Ketamine
A defining feature of ketamine’s dissociated brain state is the uncoupling of sensory input from conscious awareness, causing the brain to shift into an internally dominant mode of perception and processing. At the neurophysiologic level, anesthetic ketamine doses are accompanied by stereotyped electroencephalogram fluctuations between delta oscillations and spindles, like those observed in slow wave sleep and with conventional anesthetics (eg, propofol and isoflurane), and desynchronized periods characterized by high-frequency brain activity in the gamma range (Miyasaka and Domino 1968; Akeju et al 2016). Remarkably, while synchronized delta oscillations are observed in the thalamus and cortex during ketamine anesthesia, the hippocampus exhibits an activated theta-rich activity pattern (Miyasaka and Domino 1968; Weingarten 1972). This divergence between thalamocortical and limbic system activity may contribute to the distinctive clinical profile of dissociative anesthesia—marked by unresponsiveness to external stimuli alongside vivid hallucinations. At subanesthetic ketamine doses, hippocampal and entorhinal activity is disrupted such that an animal’s location during a navigation task can no longer be decoded (Masuda et al 2023).
At subanesthetic doses, the effect of ketamine on brain oscillations is more subtle and variable. One consistent finding is the decrease in the alpha oscillations, most reliably observed over the occipital cortex (Hong et al 2010; de la Salle et al 2016; Vlisides et al 2018; Dávila et al 2025). Interestingly, mechanistically distinct serotonergic psychedelics are also associated with a decrease in occipital alpha oscillations (Kometer et al 2013; Valle et al 2016). Since alpha oscillations in the visual cortex are thought to regulate sensory processing by inhibiting irrelevant or distracting input, their attenuation by mechanistically distinct agents may contribute to visual hallucinations. Ketamine and classic serotonergic psychedelics also induce similar changes in functional brain connectivity, despite distinct molecular mechanisms of action (Dai et al 2023). These neurophysiologic signatures reflect a transient reorganization of brain network topology, where default mode and salience networks may become hyperactive while sensory and executive control circuits are functionally disconnected (Menon 2011).
Because ketamine disrupts sensory perception, the effects of ketamine on sensory evoked potentials have been a subject of intense interest and investigations. While the results are somewhat varied, the general trend is that ketamine tends to preserve early aspects of evoked potentials (P1, N1) and diminish the later components (P2, N2, P3) thought to be related to information processing and the detection of patterns of stimuli (Schwertner et al 2018). Interestingly, these later components of the evoked potential are related to the feedback signals that propagate from higher-order thalamic and cortical areas toward primary sensory areas (Woodman 2010). Recent work used direct cortical recordings to probe the nature of the feedforward and feedback signals elicited in the mouse cortex by simple visual stimuli. The results strongly suggest that the overall structure of the visual evoked potential in mice is composed of 2 interacting traveling waves. The feedforward wave is initiated in the granular layer of the primary visual cortex and propagates toward higher-order cortical areas. In contrast, the feedback cortical waves propagate in the opposite direction and involve the supra- and infragranular layers (Aggarwal et al 2022). Consistent with primate recordings, the feedforward wave oscillates at a much higher frequency (~30 Hz) than the feedback wave (3–6 Hz; van Kerkoerle et al 2014). Consistent with the suppression of later aspects of the evoked potential by subanesthetic ketamine in humans, ketamine suppressed the feedback wave evoked by visual stimuli in mice (Aggarwal et al 2024). Interestingly, under ketamine, spontaneous activity in the absence of visual stimuli closely resembled feedback waves elicited by visual stimuli during normal wakefulness (Aggarwal et al 2024). This spontaneous emergence of activity patterns that are normally engaged in processing of visual information may form the neurophysiologic basis of sensory hallucinations that accompany ketamine administration. Remarkably, the feedback waves identified by Aggarwal et al (2024) were essentially identical to those observed by Vesuna et al (2020) in mice and human subjects.
Cortical activity evoked by transient magnetic stimulation (TMS) in the presence of dissociative and conventional anesthetics has received much attention. During wakefulness, TMS-evoked responses exhibit complex and spatially extensive activity patterns; however, during slow wave sleep and brain injury and in the presence of conventional anesthetics, evoked responses are significantly simplified (Massimini et al 2005; Ferrarelli et al 2010; Sarasso et al 2014; Sarasso et al 2015). Interestingly, in a ketamine-induced state and in REM sleep (a state associated with vivid dreaming), the responses are essentially similar to those observed during wakefulness (Massimini et al 2010; Sarasso et al 2015).
How can one explain the fact that potentials evoked by naturalistic sensory stimuli are attenuated under ketamine, while those evoked by magnetic pulses remain similar to those observed in the waking brain? Investigations of the specific circuits engaged by TMS are beginning to shed some light on this issue. TMS pulses preferentially suppress calcium signals in the apical dendrites of layer 5 neurons—an effect presumed to be mediated by local layer 1 interneurons (Murphy et al 2016). Apical dendrites of L5 neurons are the nexus where feedback and feedforward sensory streams converge (Larkum 2013; Bachmann et al 2020; Storm et al 2024). In contrast to L5 neurons, whose soma are not excited by TMS, L2/3 neurons are robustly activated. Thus, the essential distinction between sensory evoked potentials and those evoked by TMS under ketamine may arise because TMS probes a subset of the normally integrated feedforward and feedback pathways. Combined with the findings of Aggarwal et al 2022 and 2024, these results suggest that ketamine induces dissociation by selectively suppressing the interplay between feedforward pathways that supply specific stimulus information and the feedback pathways that supply context and salience to the stimuli.
The ability of TMS pulses to evoke complex responses has been linked to statistical criticality in brain activity—a property reflecting the tendency of neural networks to operate near a critical point between order and randomness (Maschke et al 2024). In states where statistical criticality signatures are present, TMS responses are typically complex, but in states such as those induced with conventional anesthetics, criticality signatures and complexity of TMS responses are attenuated (Maschke et al 2024). Here again, ketamine is a clear outlier to conventional anesthetics: multiple studies converge on the fact that the brain exhibits statistical criticality even when the subject is rendered unresponsive by ketamine (Maschke et al 2024). While the presence of statistical criticality under ketamine may explain why the ketamine-induced state is rich in sensory experience, it is not clear how this hypothesis accounts for the fact that sensory responsiveness is greatly attenuated and the contents of conscious experience are dissociated from sensory reality. Dávila et al (2025) recently addressed this issue by focusing on a distinct, dynamical notion of criticality. Consistent with previous work, they demonstrate that during normal wakefulness, cortical dynamics are observed predominantly at the critical boundary between damped and exploding oscillations (Alonso et al 2014; Solovey et al 2015; Tagliazucchi et al 2016; Toker et al 2022). Dávila et al showed that with increasing doses of ketamine, as dissociative symptoms become more and more intense, the dynamics become progressively stabilized, especially in higher-frequency ranges. Thus, it may turn out that dynamical measures of criticality depend on the intact interplay between feedforward and feedback pathways, while statistical criticality measures the capacity for conscious experience without requiring this experience to be correlated to sensory stimuli. While these are highly intriguing findings, it should be noted that statistical and dynamical measures of criticality are inferred from highly indirect measures of brain activity, which rely on empirical models that await further mechanistic evaluation.
Untangling Ketamine Dissociation from Antidepressant Responses
Whether ketamine’s dissociative side effects are necessary for its antidepressant effects remains a matter of debate (Ballard and Zarate 2020). Luckenbaugh et al (2014) examined whether the side effects of ketamine, such as dissociation, psychotomimetic symptoms, or hemodynamic changes, are related to its antidepressant efficacy in treatment-resistant depression. Data from 108 inpatients with major depressive or bipolar disorder were analyzed following a single subanesthetic ketamine infusion. The results showed that greater dissociation, as measured by Clinican-Administered Dissociation States Scale (CADSS) at 40 min, significantly predicted greater and more sustained antidepressant effects on Hamilton Depression Rating Scale (HDRS) scores at 230 min and day 7. In contrast, psychotomimetic symptoms, manic symptoms, and vital sign changes were not correlated with antidepressant response. These findings suggest that dissociation during infusion may serve as a predictive marker for ketamine’s antidepressant efficacy.
In a dose-response study, Fava et al (2020) evaluated the antidepressant efficacy of various subanesthetic intravenous ketamine doses (0.1, 0.2, 0.5, and 1.0 mg/kg) versus active placebo (midazolam) in treatment-resistant depression. Among 99 outpatients, the standard dose (0.5 mg/kg) and high dose (1.0 mg/kg) produced significantly greater antidepressant effects as compared with placebo, especially at day 1, while lower doses showed no consistent benefit. Importantly, dissociative symptoms were more prominent at higher ketamine doses (0.5 and 1.0 mg/kg) and paralleled the doses that produced the clearest antidepressant response. This study supports the observation that greater dissociation is associated with more robust antidepressant effects, consistent with emerging evidence linking ketamine’s subjective effects (eg, dissociation) to its therapeutic efficacy.
If ketamine-induced subjective experiences are thought to play a key role in the therapeutic actions of rapid-acting antidepressants, general anesthesia offers a setting in which patients can be rendered unconscious and effectively masked to the psychedelic component of treatment. This provides a straightforward way to examine drug-induced brain changes in the absence of conscious experience. The PODCAST trial, an international randomized study of 670 patients aged ≥60 y undergoing major surgery, tested these effects by administering either intraoperative subanesthetic ketamine (0.5 or 1.0 mg/kg) or placebo (Mashour et al 2018). On postoperative day 3, depression rates were similar between placebo and ketamine groups. Overall, depressive symptoms rose from 9.6% preoperatively to 16.6% on day 3, then declined to 11.9% by day 30; about half of affected patients had no prior depression history. The authors concluded that ketamine did not prevent or improve depressive symptoms after surgery. More recently, Lii et al (2023) conducted a triple-masked, randomized, placebo-controlled trial in 40 adults with major depressive disorder undergoing routine surgery, in which participants received either intravenous ketamine (0.5 mg/kg) or saline placebo during general anesthesia. Depression severity, as measured by Montgomery-Asberg Depression Rating Scale over the first 3 postoperative days, did not differ significantly between groups. Only 36.8% correctly guessed their treatment allocation, indicating successful masking. The study concluded that ketamine administered under surgical anesthesia provided no greater short-term antidepressant benefit than placebo.
To further explore whether ketamine’s dissociative experiences and rapid antidepressant effects depend on wakefulness, Markman et al (2024) studied chronically stressed mice and found that coadministration of isoflurane abolished ketamine’s dissociative-like behaviors (Figure 6A–E) and suppressed the characteristic neuronal activity patterns in prefrontal pyramidal neurons typically seen during ketamine exposure (Figure 6H and I). These changes coincided with the loss of antidepressant-like behavioral effects (Figure 6F) and induction of c-Fos, a marker of activity-dependent plasticity (Figure 6I). Together, the findings suggest that wakefulness and conscious experience may be critical for ketamine’s rapid antidepressant actions and that masking these experiences with anesthesia could disrupt key plasticity mechanisms underlying therapeutic benefit (Figure 6J).
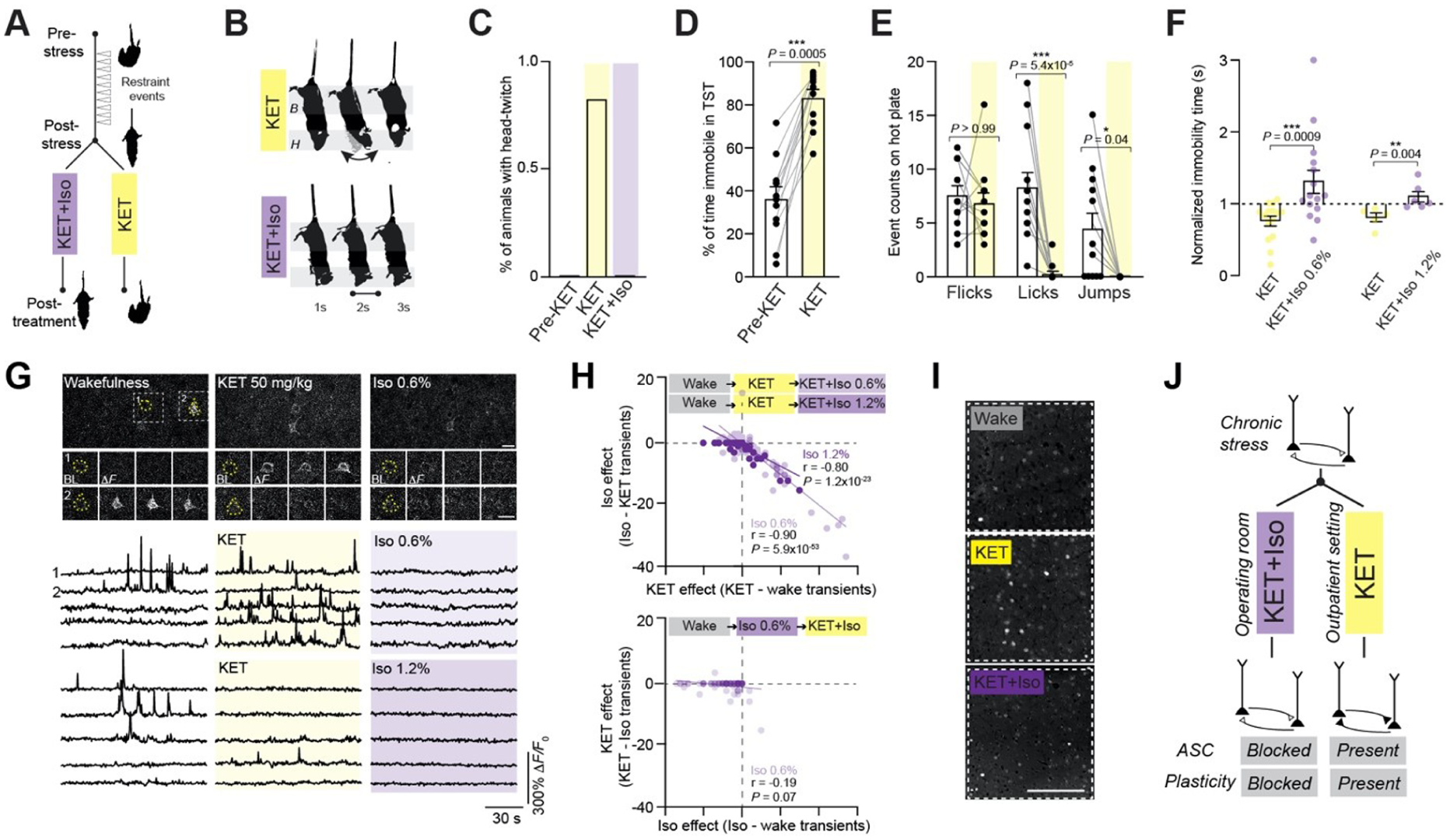
Coadministration of ketamine (KET) and isoflurane (Iso) blocks KET’s head twitch, changes in neuronal activity, and ensuing antidepressant-like response. (A) Schematic of chronic stress protocol and treatment design. Mice start protocol with more mobility in tail suspension and transition toward more immobility. Antidepressant-like activity was assessed after mice received either a single dose of KET alone (50 mg/kg intraperitoneal) or KET with Iso (0.6% [sedation] or 1.2% [anesthesia]). (B) Representative behavioral response: H, shaded region denotes location of head twitch; B, body remains stable. (C) Percentage of animal’s head-twitch response in tail suspension test (TST) under KET alone and KET with Iso. KET induced a continuous vertical head twitch (curved arrow), whereas KET with Iso eliminated this effect (straight line). (D) Percentage of time spent immobile in TST before and after KET injection. (E) Event counts for hot plate–associated behaviors—flicks, licks, and jumps—before and after KET. (F) Effect of KET alone or KET with Iso 0.6% or 1.2% on TST immobility time 24 h following treatment. (G) Top, 2-photon images of L2/3 neurons from prefrontal (Cg1) region from Thy1-GCaMP6f mouse under wakefulness, KET 50 mg/kg, and Iso 0.6%. Two pyramidal neurons are outlined with yellow dashed circles in left image. Wake-inactive cell activates after KET (cell 1). Wake-active cell inactivates after KET (cell 2). No activity in cell 1 or 2 under Iso state. Baseline and multiple peak ΔF/F0 images during different states are shown for cells 1 and 2. Scale bar, 20 µm. Bottom, Representative GCaMP6 traces of individual neurons under wakefulness, KET, and Iso. Note the activity suppression induced by Iso. (H) Difference in calcium transients induced by KET from baseline wakefulness vs Iso from KET state in all L2/3 cells recorded. Note the strong negative correlation between activity induced by KET and Iso. When Iso was administered before KET (lower graph), KET’s reconfiguration of activity was blocked. (I) Representative Cg1 regions from chronically stressed mice immunostained for c-Fos under wakefulness, KET, and KET with Iso. Note that KET with Iso blocked a KET-induced increase in c-Fos. (J) Schematic shows circuit dysfunction in chronic stress where KET treatment in the absence of general anesthesia induces an altered state of consciousness (ASC) and activity-dependent neuroplasticity. The combined effects likely contribute to KET’s rapid-acting antidepressant actions. Reproduced with permission from Markman et al (2024), Psychedelic Medicine.
However, a recent systematic review evaluated the broader literature on whether ketamine’s dissociative and psychotomimetic effects are consistently correlated with antidepressant outcomes in patients with major depressive disorder (Mathai et al 2020). Eight studies were selected from 556 screened reports, focusing on trials that used single-dose ketamine and measured subjective effects with validated scales (CADSS, the Brief Psychiatric Rating Scale (BPRS), 5-Dimensional Altered States of Consciousness Rating Scale (5D-ACS) alongside depression outcomes. The review found that 37.5% of studies reported a significant correlation between subjective effects (primarily dissociation) and antidepressant response, although the majority of studies either did not identify a correlation or did not systematically examine it. The authors concluded that this relationship remains unclear and underexplored and emphasize the need for future research to better define the potential role of subjective psychedelic-like effects in ketamine’s therapeutic efficacy—similar to emerging frameworks in MDMA and psilocybin research and therapy.
Potential Enduring Impacts of a Single Dissociative Dose on Neuronal Function
Preliminary correlative evidence suggests that ketamine’s rapid and sustained antidepressant effects arise not only from its well-characterized receptor-level pharmacology but also from epigenetic modifications that reprogram neural circuits to promote long-term plasticity (Inserra et al 2024). Upon administration, ketamine induces a cascade of molecular events, beginning with open NMDA receptor blockade and leading to enhanced glutamate release and subsequent AMPA receptor activation (Zorumski et al 2016; Johnston et al 2024; Lucantonio et al 2025; Figure 4, bottom panel). At the same time, ketamine can directly bind to TrkB, thereby facilitating synaptic localization of TrkB and its activation by BDNF (Casarotto et al 2021; Ma et al 2025). This step triggers downstream signaling pathways such as mTOR activation (Aguilar-Valles et al 2021), which in turn promotes the transcription of plasticity-related genes (Dwyer and Duman 2013). A critical mechanism underlying this transcriptional reprogramming is likely rapid histone modification, altering gene expression patterns. Ketamine increases histone H3 and H4 acetylation at promoter regions of genes such as BDNF, cFos (cellular Fos), and Arc (activity-regulated cytoskeleton-associated protein), which opens up chromatin structure and allows for increased gene expression (Shi et al 2020; Inserra et al 2024). This is partly mediated by the suppression of histone deacetylase activity, either directly or via secondary signaling molecules (Autry et al 2011; Ribeiro-Davis et al 2024). Additionally, ketamine alters histone methylation patterns, potentially reducing repressive marks such as H3K27me3, further contributing to gene activation (Figure 3C). Parallel to histone changes, ketamine affects DNA methylation (Dawson et al 2024; Wellington et al 2025), for example, by reducing methylation at the BDNF promoter in the prefrontal cortex and hippocampus (Krystal et al 2019). This demethylation is thought to stem from decreased activity of DNA methyltransferases, allowing previously silenced genes to become transcriptionally active again (Nepton et al 2024; Semple et al 2025).
Furthermore, ketamine modulates noncoding RNAs and miRNA pathways, which regulate the stability and translation of mRNAs involved in synaptic plasticity and neuronal survival (Fitz-James and Cavalli 2022; Inserra et al 2024). Through these combined epigenetic mechanisms, ketamine shifts the transcriptional landscape of key brain regions such as the prefrontal cortex, hippocampus, nucleus accumbens, and amygdala (Abdallah et al 2017). These modifications lead to increased dendritic spine growth (Li et al 2010), enhanced synaptogenesis (Duman and Aghajanian 2012), and circuit remodeling that persist well beyond the drug’s acute pharmacokinetic window (Moda-Sava et al 2019). Importantly, this epigenetic reprogramming provides a mechanistic basis for the prolonged antidepressant effects of ketamine (Kawatake-Kuno et al 2021).
The epigenetic consequences of dissociative ketamine doses remain essentially uncharted. We propose that dissociative dosing augments the pharmacodynamic mechanisms engaged at antidepressant levels with additional dose-dependent epigenetic processes that can rapidly and enduringly reconfigure neural circuit function (Inserra et al 2024; Lewis et al 2024). Dissociative doses of ketamine likely induce broader and more intense epigenetic reprogramming, potentially affecting distinct neuronal populations or gene networks, such as large-scale disruptions of cortical and thalamocortical connectivity, that extend beyond circuits directly related to mood regulation (Figure 3C). Epigenetic modulation may consolidate or stabilize these network-level changes by altering the transcriptional programs that regulate GABAergic interneuron function (Kawatake-Kuno et al 2024; Fogaça et al 2025), NMDA receptor subunit composition (Villéga et al 2024), or synaptic scaffolding proteins (Kim et al 2023), resulting in sustained alterations in neural synchrony, sensory gating, and the integration of conscious experience. Furthermore, emerging evidence indicates that neurotransmitters and neuromodulators can become covalently incorporated into diverse histone and other proteins via enzymatic and nonenzymatic posttranslational modifications (Kim et al 2023). This raises the intriguing possibility that dissociative ketamine doses may produce drugylation—the covalent attachment of drugs to proteins with yet unknown consequences for neuronal function (Geib et al 2021).
Clinical Implications and Future Directions
Understanding ketamine’s cortical mechanisms has substantial clinical relevance. Together, the mechanisms converge on a unifying model: ketamine induces dissociation by disrupting the balance between excitation and inhibition across cortical layers and between cortical and subcortical structures. Disinhibition of pyramidal neurons, loss of thalamocortical fidelity, and desynchronization of cortical rhythms all contribute to a decoupling of sensory input from perceptual integration (Mashour and Hudetz 2018). Beyond dissociation, these dynamics underlie its antidepressant efficacy, which may rely on a transient destabilization of cortical circuits, followed by epigenetic reprogramming and synaptic remodeling (Mathew and Zarate 2016; Abdallah, Sanacora, et al 2018). The dissociative state may thus serve not as a side effect but as a biomarker or requirement for therapeutic response. Future research should clarify how ketamine enantiomers, administration routes, and structurally related analogs modulate cortical circuit dynamics over the full spectrum of clinically and experimentally relevant doses. Tools such as genetically encoded voltage and calcium indicators, biosensors sensitive and specific to ketamine, and cell type–specific perturbation techniques will be crucial for refining mechanistic models. In addition, multimodal imaging, computational modeling, and human intracranial recordings may bridge the translational gap, linking rodent mechanisms to subjective experience and clinical outcomes. By precisely mapping how dissociation unfolds across cortical cell types, layers, and regions, we may uncover a shared neural signature for altered states of consciousness, with applications ranging from anesthesia to psychedelic therapy.
Conclusion
Ketamine’s dissociative properties arise from a complex interplay of NMDA receptor and HCN1 antagonism, interneuron-mediated disinhibition, thalamocortical decoupling, and layer-specific cortical modulation. These mechanisms collectively produce a unique neurophysiologic state that provides insights into the architecture of consciousness and points toward novel strategies for treating neuropsychiatric disorders. Ongoing investigations into these cortical processes hold promises for optimizing ketamine-based therapeutics and understanding the neural basis of dissociation.
Footnotes
Funding
Work was supported by the National Institutes of Health (NIH) R35GM151160-01 and American Society of Regional Anesthesia Chronic Pain Medicine Research grant to J.C., NIH R01GM151556 to A.P., the Della Martin Foundation to L.L., and the Howard Hughes Medical Institute to L.L.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.