
Abstract
Background
Methadone is a unique µ-opioid receptor agonist. Although several researchers have insisted that the pharmacological effects of methadone are mediated through the blockade of NMDA receptor, the underlying mechanism by which methadone exerts its distinct pharmacological effects compared to those of other µ-opioid receptor agonists is still controversial. In the present study, we further investigated the pharmacological profile of methadone compared to those of fentanyl and morphine as measured mainly by the discriminative stimulus effect and in vitro assays for NMDA receptor binding, µ-opioid receptor-internalization, and µ-opioid receptor-mediated β-arrestin recruitment.
Results
We found that fentanyl substituted for the discriminative stimulus effects of methadone, whereas a relatively high dose of morphine was required to substitute for the discriminative stimulus effects of methadone in rats. Under these conditions, the non-competitive NMDA receptor antagonist MK-801 did not substitute for the discriminative stimulus effects of methadone. In association with its discriminative stimulus effect, methadone failed to displace the receptor binding of MK801 using mouse brain membrane. Methadone and fentanyl, but not morphine, induced potent µ-opioid receptor internalization accompanied by the strong recruitment of β-arrestin-2 in µ-opioid receptor-overexpressing cells.
Conclusions
These results suggest that methadone may, at least partly, produce its pharmacological effect as a β-arrestin-biased µ-opioid receptor agonist, similar to fentanyl, and NMDA receptor blockade is not the main contributor to the pharmacological profile of methadone.
Background
µ-opioid receptor agonists have been used for the treatment of patients who are experiencing moderate to severe cancer pain or non-cancer pain such as chronic inflammatory and postoperative pain. However, µ-opioid receptor agonists produce several side effects, such as emesis, constipation, drowsiness, hallucination, and delirium. Particularly, constipation is a distressing side effect of opioids that are used to manage pain and occurs in 40% to 95% of patients treated with opioids. 1 Severe side effects induced by µ-opioid receptor agonists could limit their use, which may have serious consequences, such as uncontrolled pain. Therefore, reducing such effects is an important strategy for improving the quality of life of patients suffering from pain. 2
Morphine, oxycodone, fentanyl, and methadone are clinically prescribed opioids that have prominent antinociceptive effects. These µ-opioid receptor agonists have distinct pharmacological profiles by which they exert their antinociceptive effects through different regions of the central nervous system.3,4 Among these µ-opioid receptor agonists, methadone is known to be useful in patients with pain that does not respond to other analgesics. 5 Methadone is considered to have unique and diverse pharmacologic properties, including NMDA receptor antagonism, inhibition of serotonin and noradrenaline uptake, and affinity for δ-opioid receptors, in addition to µ-opioid receptor agonistic action.6,7 In fact, NMDA receptor antagonists could enhance the antinociceptive effects of morphine in rats. 8 However, there is not enough evidence regarding whether the pharmacological action of methadone involves the blockade of NMDA receptors. If the blockade of NMDA receptor antagonism plays a role in the antinociceptive effects of methadone, the likelihood of inducing side effects would be lower than with other µ-opioid receptor agonists. 9
On the other hand, methadone itself is associated with less abuse potential than either morphine or heroin,10,11 and treatment with methadone has been considered to be the most effective approach as part of a comprehensive treatment program for opioid-dependent individuals. To better understand the distinct pharmacological profile of methadone, we examined the similarities and/or differences between methadone and other µ-opioid receptor agonists with respect to discriminative stimulus effects and the possible involvement of the blockade of NMDA receptors in the discriminative stimulus effects of methadone. Recent studies have shown that µ-opioid receptor agonists have different efficacies and pathways for the activation of coupled G-proteins and induce the endocytosis of µ-opioid receptor receptors, which may reflect their signaling effects.12,13 Therefore, we also investigated the relative ability of methadone to promote µ-opioid receptor internalization and β-arrestin recruitment compared to fentanyl and morphine.
Methods
Animals
Male institute of Cancer Research (ICR) mice (20–25 g) (Tokyo Laboratory Animals Science Co. Ltd., Tokyo, Japan) and male Fischer 344 rats 200–230 g (Charles River Japan Inc., Atsugi, Japan) were used. Food and water were available ad libitum in their home cages except in the case of drug discrimination study. Mice and rats were housed in a room maintained at 22 ± 1℃ with a 12-hr light-dark cycle (light on 8:00 a.m. to 8:00 p.m.). The present study was conducted in accordance with the Guiding Principles for the Care and Use of Laboratory Animals at Hoshi University, as adopted by the Committee on Animal Research of Hoshi University. Every effort was made to minimize the numbers and any suffering of animals used in the following experiments.
Hot-plate test
The antinociceptive response was evaluated by recording the latency to paw licking or tapping in the hot-plate test (55 ± 0.5℃; Muromachi Kikai Co., Ltd., Tokyo, Japan) as described previously. 14 To prevent tissue damage, we established a 30-s cut-off time. Antinociceptive effects were measured after administration of methadone (1.0–10 mg/kg s.c.). Each animal served as its own control, and the latency to a response was measured both before and after drug administration. Antinociception was calculated as a percentage of the maximum possible effect (% antinociception) according to the following formula: % antinociception = (test latency − predrug latency)/(cut off time − predrug latency) × 100. The antinociceptive response represents the mean ± SEM of the % antinociception.
Gastrointestinal transit
Gastrointestinal transit was conducted based on previous method. 15 Briefly, mice were fasted for 12 hr before the experiments. At 10 min after the methadone (1.0–10 mg/kg s.c.) injection, blue ink (0.3 ml/mouse; Pilot Co. Ltd., Tokyo, Japan) was administered orally; 30 min after the administration of blue ink; the animal was killed by cerbical dislocation, and the small intestine was removed. The percentage inhibition of gastrointestinal transit was calculated as follows: (distance traveled by the ink/length from the pylorus to the cecum) × 100.
Effect on colonic expulsion
The effects of methadone on colonic propulsion were evaluated as described previously. 9 Briefly 10 min after the administration of methadone (1.0–10 mg/kg s.c.), a glass bead (3 mm in diameter; BZ-3 Ikeda Rika, Tokyo, Japan) was inserted into the distal colon to a depth of 2 cm from the anus with a silicone tube (2 mm in diameter). The time required to expel the bead was measured up to 120 min.
Drug discrimination study
Discrimination training was performed according to the method described previously. 16 Briefly, before they were trained to discriminate between methadone and saline, all of the rats were trained to press a lever. Training began under a reinforcement schedule of fixed ratio 1 (FR 1) in which the rat was presented with a food pellet each time it pressed a lever. When reinforcement was provided, the light above the lever was illuminated. The FR requirement for food reinforcement was gradually increased to a value of 10. Rats were trained to discriminate between 2.0 mg/kg of methadone (s.c., 30 min) and saline. In the discrimination training, training drugs (D) or saline (S) were administered in a session-to-session sequence of DDSS (double alternation schedule), and the assignment of left and right levers to drug and saline states was counterbalanced. The rats were required to respond on the stimulus-appropriate lever to obtain reinforcement; there were no programmed consequences for responding on the incorrect lever. Substitution tests were only performed after the discrimination criterion described later had been satisfied for at least five consecutive daily discrimination training sessions (accuracy of at least 83% and fewer than 12 responses to obtain the first reinforcement [FRF]).
After the animals attained the criterion, generalization tests were initiated; test sessions were performed after the discrimination criterion described earlier had been satisfied for at least three consecutive sessions. If accuracy criteria were not reached during the training session, rats were trained until their accuracy was maintained for minimum of three training sessions. During the test session, the rats were placed in the operant box until they had made 10 responses on either lever or 5 min had elapsed. The pretreatment time and doses of drugs used were 30 min for morphine (1.0–5.6 mg/kg, s.c.), MK-801 (0.01–0.1 mg/kg, i.p.); and 15 min for fentanyl (0.01–0.03 mg/kg, s.c.). Drugs were considered to have generalized to the discriminative stimulus effects of the training drugs if more than 80% of the responses were on the drug-appropriate lever. If the rats did not make 10 responses during each test session, the response was judged to have been disrupted.
Receptor binding assay
The mouse forebrain was weighed and homogenized in a 10-fold volume of 50 mmol/L ice-cold Tris-HCl buffer (pH 7.4). After centrifugation at 50,000 g for 20 min at 4℃, the pellets were washed once with a 10-fold volume of buffer and centrifuged under the same conditions. The resulting pellet was resuspended and used as the membrane fraction (20 mg tissue/ml). The binding assay was performed in triplicate with [3H] DAMGO at 2 nM, [3H]MK-801at 3 nM in a final volume of 500 µl that contained 50 mM Tris-HCl buffer, pH 7.4, and 200 ml homogenized membrane fraction. After 60-min incubation at 25℃, the mixture was trapped on a GF/C glass fiber filter. Specific binding for µ-opioid receptor and NMDA receptor were defined as the difference in binding observed in the absence and presence of 1 mM unlabeled DAMGO and MK-801. Radioactivity in the samples was determined with a liquid scintillation analyzer. All receptor binding curves were fitted using Prism software (version 5.0 a; GraphPad Software, La Jolla, CA).
Internalization assay
For the receptor internalization assay, we obtained cDNA for N-terminal Halotag®-fused human µ-opioid receptors from Kazusa DNA Research Institute (Kisaragi, Chiba, Japan). The clone was stably expressed in human embryonic kidney (HEK) 293 cells, and these cells were used for the receptor internalization assay. Cells were washed with Krebs-Ringer HEPES buffer, then stained with 0.5 µM Halotag® Alexa 488, which is a membrane-impermeable dye for Halotag® by binding irreversibly, for 15 min. Stained cells were washed with Krebs-Ringer HEPES and then stimulated with several µ-opioid receptor ligands for 60 min. Intensities of pixels of green-stained Halotag-µ-receptors before and after stimulation of several ligands were captured by a confocal microscopy (Carl Zeiss Japan, Tokyo, Japan). The ratio of internalized receptors was analyzed quantitively.
Quantitative analysis of receptor internalization
The levels of Halotag-µ-receptor internalization were quantified as follows: the receptor internalization was quantified using single confocal image that includes the nucleus and a large area of cytoplasm.
A line was drawn along the outside of the cell and total cell fluorescence (cell membrane + cytoplasm) was measured as the total intensities of pixels with fixed cell preparation. In order to measure the cell membrane and cytoplasmic receptor separately, a second line was drawn inside the cell membrane 2 µm from the first line, and the intensities of pixels at the same fluorescence density inside the second line were measured. The percentage of fluorescence in the cytoplasm was calculated from the fluorescence intensities in the cytoplasmic region (second measure pixels) divided by the total fluorescence intensities (first measure pixels). Δ% internalization was quantified according to previous paper. 17 Briefly, fluorescence intensities obtained from 15 to 20 cells were calculated as an independent experiment.
β-arrestin-2 recruitment
The PathHunter enzyme complementation assay (DiscoveRx) was performed according to the manufacturer’s protocol and previous report 18 and detect for chemiluminescence on a GloMax-Multi Detection System (Promega Co., WI, USA). Briefly, when β-arrestin-2 translocates to active receptor, the complementary β-galactosidase fragments fused to receptor and β-arrestin-2 interact to form a functional enzyme.
Drugs
The drug used in the present study was dl-methadone (Teikoku Seiyaku Co., Ltd, Kagawa, Japan), morphine hydrochloride (Daiichi-Sankyo Co., Ltd, Tokyo, Japan), fentanyl citrate (Hisamitsu Pharmaceutical Co Inc, Tokyo, Japan), and MK-801 (Sigma-Aldrich Co.). All drugs were dissolved in saline and administered in a volume of 10 and 1 ml/kg to administer for mice and rats, respectively.
Statistical analysis
Data are expressed as the mean ± SEM of six to eight animals. The statistical significance of differences between groups was assessed by the Mann–Whitney test. The 50% effective dose (ED50) or inhibitory concentration (IC50) values were determined using an analysis of variance and linear regression techniques. Where appropriate, a one-way analysis of variance (ANOVA) followed by the Bonferroni multiple comparisons test was used for statistical analysis. All statistical analyses were performed using Prism software (Version 5.0 a; GraphPad Software, Inc., La Jolla, CA). A P value of 0.05 was considered to reflect significance.
Results
Methadone-induced antinociception and inhibition of gastrointestinal and colorectal transit
Dose-response curves for the pharmacological effects of methadone, such as antinociception, inhibition of gastrointestinal, and colorectal transit, and the corresponding ED50 values are shown in Figure 1. Methadone dose-dependently produced antinociceptive effects as well as the inhibition of gastrointestinal and colorectal transit in mice. The potencies of methadone for inducing these pharmacological effects (ED50) were almost the same.
Dose-response curves for the antinociceptive effect and inhibitory effects on gastrointestinal transit (GIT) and colonic expulsion induced by methadone in mice. Groups of mice were treated with methadone (1.0–10 mg/kg s.c.). Antinociceptive effects and the inhibition of GIT or colonic expulsion were measured at 10 minutes after the subcutaneous injection of methadone. Antinociceptive effects and inhibition of GIT or colonic expulsion are expressed as the % effect. The data represent the mean ± S.E.M. of five to seven animals. ED50 values were determined using an analysis of variance and linear regression techniques. Values in parentheses indicate the 95% confidence range.
Discriminative stimulus effects of methadone
Rats required approximately 50 sessions to acquire methadone-saline discrimination. Once rats attained the criterion, drug-saline discrimination stabilized and was maintained with a high degree of accuracy (on average at least 90% of the responses that occurred before the first reinforcement). During the dose-response tests, methadone (0.5–2.0 mg/kg) produced a dose-related increase in drug-appropriate responses in all of the rats that had been trained to discriminate between methadone and saline without a reduction in the response rate (Figure 2a). In substitution tests, morphine (5.6 mg/kg) and fentanyl (0.017 and 0.03 mg/kg) completely substituted for the discriminative stimulus effects of methadone (Figure 2b). On the other hand, MK-801 (0.01–0.1 mg/kg) did not substitute for the discriminative stimulus effects of methadone (Figure 2c). Behavioral sedation accompanied by a decrease in the response rate was observed at the highest dose of MK-801 (0.1 mg/kg) during the substitution test.
Dose-response of methadone (a) and substitution of morphine, fentanyl and MK-801 (b) to the discriminative stimulus effects of methadone (top panel) and the response rates (bottom panel) in rats that had been trained to discriminate between 2.0 mg/kg methadone and saline. Each point represents the mean percentage of methadone-appropriate responding and the mean response rates with SEM of eight animals.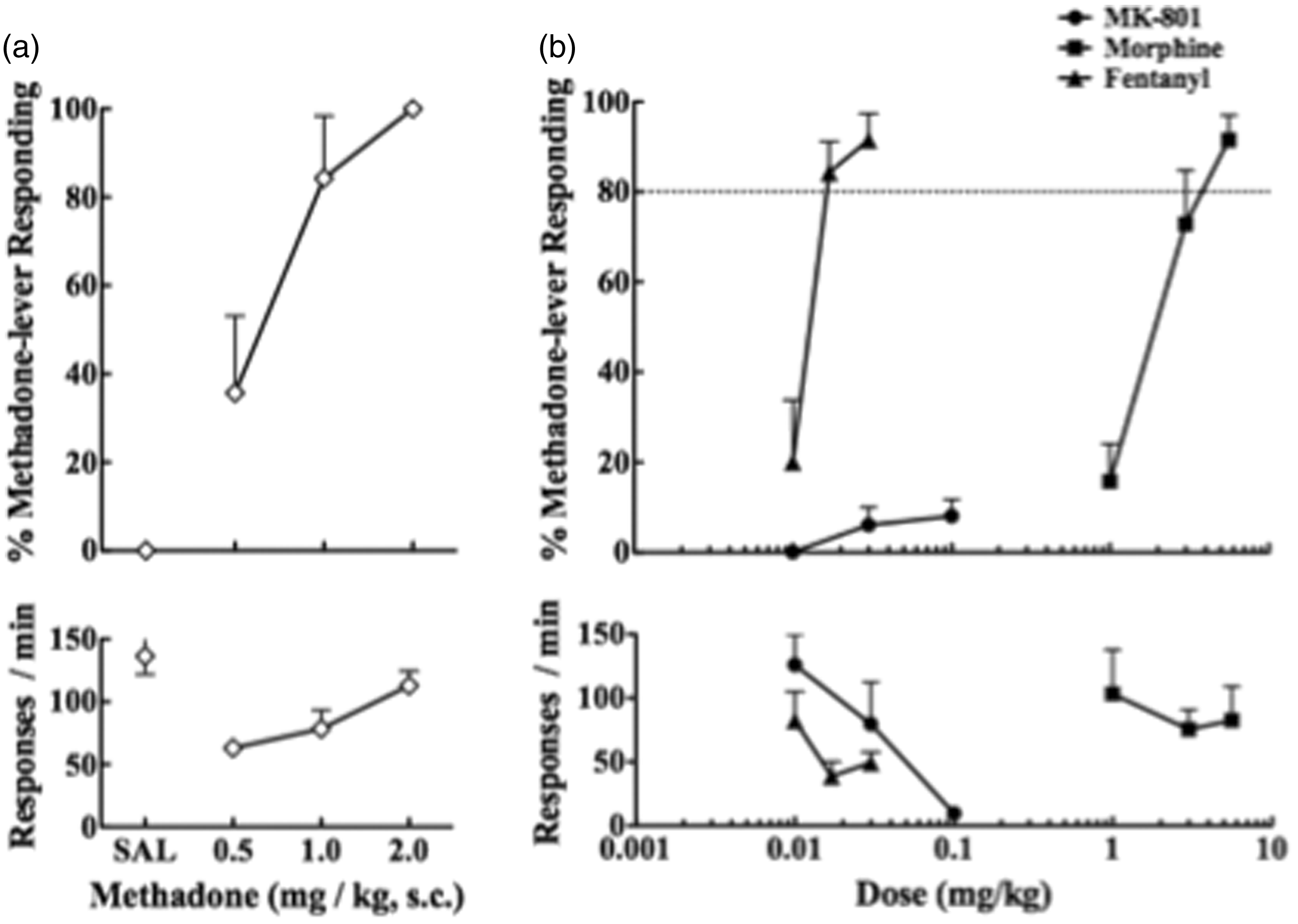
Binding properties of methadone with µ-opioid and NMDA receptors
In mouse brain membranes without the cerebellum, we determined the competitive displacement binding of [3H]DAMGO and [3H]MK-801 with graded concentrations (10−10 to 10−6) of unlabeled methadone. The binding of [3H]DAMGO was displaced by methadone in a concentration-dependent manner with an IC50 value of 10−7.977 M. On the other hand, the binding of [3H]MK-801 was scarcely displaced by methadone, even with the use of 1 mM methadone (Figure 3).
Displacement of the binding of the µ-opioid receptor ligand [3H] DAMGO or NMDA receptor ligand [3H] MK-801 in membranes of mouse brain without the cerebellum by methadone. Experiments were performed in the presence of [3H] DAMGO (2 nM) or [3H] MK-801 (3 nM) and increasing concentrations of methadone. The data represent the mean ± SEM of three to four samples. The IC50 values were determined using an analysis of variance and linear regression techniques. To calculate the IC50 values, at least five drug doses were used, and three samples were used for each dose. Values in parentheses indicate the 95% confidence range.
Methadone-induced internalization of μ-opioid receptor
We constructed N-terminal-Halo-tagged µ-opioid receptors that were stably overexpressed in HEK-293 cells. Halo fluorescence was observed mainly in the cell membrane and scarcely in the cytoplasm of HEK-293 cells (data not shown). As shown in Figure 4, morphine (10 µM) did not significantly affect the distribution of µ-opioid receptors in HEK-293 cells. In contrast, fentanyl (1 µM) as well as methadone (10 µM) significantly translocated µ-opioid receptors from the cell membrane to the cytoplasm within HEK-293 cells. The potencies for internalizing µ-opioid receptors were in the order: fentanyl = methadone » morphine.
Localization of µ-opioid receptors after activation by µ-opioid receptor agonists in HEK-293 cells that stably overexpressed Halo-µ-opioid receptors. Δ% of internalization of µ-opioid receptors after the administration of morphine, methadone, and fentanyl. Blue: Hoechst nuclear staining, Green: Halo-µ-opioid receptors. Each column represents the mean with SEM of three independent experiments. ***p < 0.001 vs. buffer group.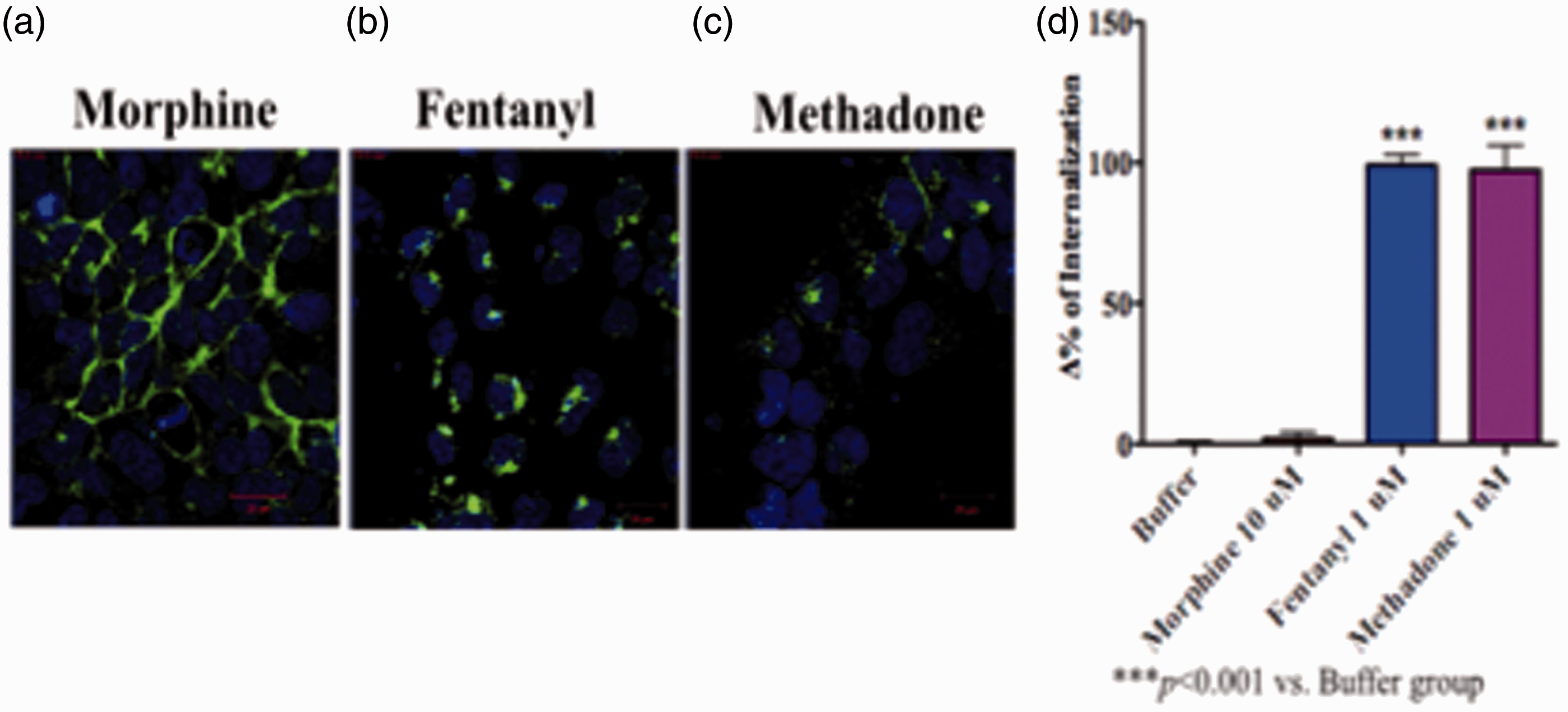
Methadone-induced β-arrestin-2 recruitment
Treatment with morphine (10 µM), fentanyl (1 µM), and methadone (10 µM) produced a dose-dependent increase in luminescence that reflected β-arrestin-2 recruitment (Figure 5). Particularly, fentanyl and methadone robustly increased luminescence signals with a typical sigmoid curve. On the other hand, morphine produced only a weak increase in luminescence.
Measurements of β-arrestin-2 recruitment by the PathHunter enzyme complementation assay in CHO cells that overexpressed human µ-opioid receptors. β-arrestin-2 recruitment was measured in terms of an increase in luminescence after the administration of morphine, methadone, and fentanyl (A). Δ% internalization of µ-opioid receptor after the administration of morphine, methadone, and fentanyl (B). Each column represents the mean with SEM of three independent experiments.
Discussion
Previous studies have demonstrated that µ-opioid receptor agonists have distinct pharmacological profiles by exerting both antinociceptive effects, as observed in several models of pain, and side effects, such as constipation and respiratory inhibition, in rodents.3,4 We previously showed that morphine, oxycodone, and fentanyl displayed different pharmacological profiles to inhibit gastrointestinal transit and colorectal expulsion through distinct mechanisms in a region-specific manner, in comparison to their antinociceptive effects. Particularly, the potencies of these µ-opioid receptor agonists for inducing gastrointestinal/colorectal transit, as compared to their antinociceptive effects, were in the order morphine = oxycodone > fentanyl. 9 These findings are consistent with the clinical observation that morphine- and oxycodone-induced constipation is a serious problem in patients when these compounds are used to control pain, 19 whereas fentanyl may have a lower risk of producing constipation than oxycodone 20 or morphine. 21 In the present study, we found that the ED50 potency of methadone for inducing gastrointestinal/colorectal transit was comparable to that for its antinociceptive effects, which is very similar to the results with fentanyl using the exact same protocols in mice. 9
In the present drug-discriminative study, we demonstrated that morphine and fentanyl fully substituted for the discriminative stimulus effects of methadone in rats. We previously reported that the ED50 values for producing the antinociceptive effects of morphine and fentanyl were 6.6 mg/kg and 0.06 mg/kg, respectively. 9 In this study, a relatively lower dose of fentanyl (0.017 mg/kg) was required to substitute for the discriminative stimulus effects of methadone, whereas almost equal doses of morphine were needed to produce these pharmacological effects. With regard to these results, Vann et al. 22 demonstrated that at least 10 mg/kg of s.c. morphine was required to substitute for the discriminative stimulus effects of methadone in rats trained to discriminate between 3.0 mg/kg of methadone and saline, whereas i.p. morphine (0.3–10 mg/kg) did not substitute for the discriminative stimulus effects of methadone. Therefore, the pharmacological profiles for producing the discriminative stimulus effects of methadone are slightly different from those of morphine, and this difference may at least partly be related to the usefulness of methadone maintenance for the treatment of opioid dependence.
Previous receptor binding studies have demonstrated that methadone has high affinity for µ-opioid receptors and also binds to NMDA receptors with lower affinity.23,24 Currently, there is some uncertainty regarding whether the antinociceptive effects of methadone are mediated solely by its agonistic action through µ-opioid receptors or whether the possible binding of methadone to NMDA receptors may contribute to its pharmacological action including antinociceptive effects. In the present binding study, methadone replaced [3H]-DAMGO binding with relatively high affinity, whereas it failed to replace [3H]-MK-801 binding at micromolar concentrations in mouse brain membrane, indicating that methadone apparently has weaker affinity for NMDA receptors than for µ-opioid receptors. Carpenter et al. 25 reported that the antinociceptive effect of methadone was mainly suppressed by naloxone in intact rats, whereas others demonstrated that blockade of NMDA may contribute to the antinociceptive effects of methadone in models of both neuropathic and inflammatory pain.6,7 In the substitution test, we clearly found that MK-801, unlike µ-opioid receptor agonists, failed to produce any methadone-appropriate responding in rats that had been trained to discriminate between methadone and saline. The results of the present receptor binding and drug-discrimination studies provide strong evidence that the blockade of NMDA receptors may not provide the main contribution to the discriminative stimulus effects of methadone. Interestingly, it has been reported that therapeutic concentrations of methadone are not sufficient to block NMDA receptors, as measured by an electrophysiological technique. 26 Therefore, we hypothesize that the blockade of NMDA receptor may not be directly involved in the pharmacological actions of methadone.
With regard to the distinct profile of methadone, it has been reported that l-methadone has relatively high affinity for sites that inhibit the uptake of serotonin (Ki = 14 nM), in addition to its µ-opioid receptor agonistic profile.27,28 It is well known that activation of the serotonergic system contributes to the antinociceptive action of µ-opioid receptor agonists in the spinal cord. We previously demonstrated that the serotonin uptake inhibitor fluvoxamine enhanced the antinociceptive effects of morphine, whereas it significantly suppressed the inhibition of gastrointestinal transit induced by morphine in mice. 29 As mentioned above, methadone has a lower risk of producing constipation than morphine and oxycodone. The need for a laxative to suppress methadone-induced constipation was 10 times lower than that with morphine in patients who suffered from pain.30–32 Thus, activation of the serotonergic system by l-methadone may at least partly contribute to pharmacological effects induced by methadone.
The µ-opioid receptor is the most extensively studied G-protein-coupled receptor (GPCR) for producing antinociception and is a therapeutic target for pain control. Recently, the concept of a ligand bias for GPCRs, including µ-opioid receptor, whereby a ligand stabilizes subsets of GPCR conformations to engender novel pharmacological profiles, has gained increasing prominence. With regard to the independence of G-protein signaling at GPCRs, there has been a growing appreciation of β-arrestin-mediated signaling, which provides an even greater opportunity for GPCR-mediated pharmacology. The activation of β-arrestin regulates many of its downstream signaling pathways including those of Src family kinases, extracellular signal-regulated kinase 1/2 (ERK 1/2), and phosphatases. In the present study, we found that methadone as well as fentanyl potently promoted the internalization of µ-opioid receptors and recruited β-arrestin-2. In contrast, morphine scarcely induced µ-opioid receptor internalization and showed only weak β-arrestin-2 recruitment. Fentanyl, but not morphine, has been shown to induce the phosphorylation of ERK 1/2, which may trigger the internalization of µ-opioid receptor, in striatal neurons. 33 Taken together, these findings suggest that methadone may have a pharmacological profile similar to that of fentanyl and can be classified as a β-arrestin-biased µ-opioid receptor agonist. Furthermore, this may also partly explain the difference in the pharmacological profiles of methadone and morphine. In addition, we also demonstrated that repeated treatment with fentanyl, unlike in the case of morphine, produced a rapid development of tolerance to its antihyperalgesic effect in mice with sciatic nerve ligation. 34 Therefore, methadone might have a profile similar to that of fentanyl in this regard. Further studies will be needed to address this issue.
Based on the pharmacological profile of morphine in β-arrestin-2 knockout mice, a G protein-biased ligand that shows poor β-arrestin recruitment may offer increased analgesia without respiratory depression, nausea, or constipation. However, our preliminary and present studies provide contradictory evidence that β-arrestin-biased µ-opioid receptor agonists, such as fentanyl and methadone, show weak potency for producing constipation in mice and nausea in ferrets, whereas the selective G protein-biased µ-opioid receptor ligand TRV130, which stimulates nearly undetectable levels of β-arrestin recruitment, shows similar potency for producing gastrointestinal dysfunction in mice and nausea in ferrets at equianalgesic doses to fentanyl and methadone (unpublished observation). These findings raise a new hypothesis that selective G protein-biased ligands that show poor β-arrestin recruitment may not always offer increased analgesia with improved safety and tolerability compared to currently prescribed opioids and may not selectively target a clinically beneficial signaling pathway to deliver an improved therapeutic profile.
In conclusion, the present findings strongly support the idea that methadone may act as a β-arrestin-biased µ-opioid receptor agonist, like fentanyl. Furthermore, the pharmacological profile of methadone may not directly correspond to the blockade of NMDA receptors.
Footnotes
Author Contributions
Seira Doi and Tomohisa Mori contributed equally to this work.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported in part by grants for NEXT-Supported Program for the Strategic Research Foundation at Private Universities, 2014-2018, S1411019 and Grants-in-Aid for Scientific Research (C) No. 24590740 and No. 15K08686 from the Ministry of Education, Culture, Sports, Science and Technology of Japan.