
Abstract
The basal ganglia are an intricately connected assembly of subcortical nuclei, forming the core of an adaptive network connecting cortical and thalamic circuits. For nearly three decades, researchers and medical practitioners have conceptualized how the basal ganglia circuit works, and how its pathology underlies motor disorders such as Parkinson’s and Huntington’s diseases, using what is often referred to as the “box-and-arrow model”: a circuit diagram showing the broad strokes of basal ganglia connectivity and the pathological increases and decreases in the weights of specific connections that occur in disease. While this model still has great utility and has led to groundbreaking strategies to treat motor disorders, our evolving knowledge of basal ganglia function has made it clear that this classic model has several shortcomings that severely limit its predictive and descriptive abilities. In this review, we will focus on the striatum, the main input nucleus of the basal ganglia. We describe recent advances in our understanding of the rich microcircuitry and plastic capabilities of the striatum, factors not captured by the original box-and-arrow model, and provide examples of how such advances inform our current understanding of the circuit pathologies underlying motor disorders.
Keywords
Introduction: Beyond the Extrapyramidal System
Until the early 1970s, medical practitioners were being taught that the striatum (caudate and putamen nuclei) is part of the extrapyramidal motor system and exerts a “steadying influence” (Wilson 1912) on lower motor neurons via a putative descending projection (Denny-Brown 1962; Kemp and Powell 1971). As a result of research conducted over the next two decades, in what may be called the Golden Age of basal ganglia (BG) research, this vague and imprecise description was supplanted in medical and neuroscience textbooks by the direct versus indirect pathway model (also known as the “box-and-arrow” model) of BG physiology and pathophysiology (Albin and others 1989; DeLong 1990). This model organized what had been learned during this period about the gross anatomy and physiology of the BG into a comprehensive model of how two mutually antagonistic pathways gate action and how movement disorders could be conceptualized as resulting from an imbalance between them.
The Golden Age of Basal Ganglia Research and the “Box-and-Arrow” Model
The box-and-arrow model (Fig. 1A) (in which each of the various BG nuclei is represented as a box, and the synaptic connections among them as arrows) is the product of discoveries made from the mid-1970s to the early 1990s by several research groups (Alexander and others 1986; Chang and others 1981; Chevalier and Deniau 1990; DiFiglia and others 1976; Gerfen and others 1990; Graybiel and Ragsdale 1978; Kita and Kitai 1987; Parent and others 1984; Smith and Bolam 1990). These findings revealed novel and previously unknown principles of the BG’s structure and function. The most influential finding pertained to the striatum, the main input structure of the BG. Anatomical tracing studies revealed that GABAergic spiny projection neurons (SPNs), which, in rodents, account for over 95% of striatal neurons, could be divided into two equally sized populations based on their axonal projections (Chang and others 1981; Kawaguchi and others 1990; Parent and others 1984; Penny and others 1986). The axonal arborizations of the first population projected to one or both of the output structures of the BG: the substantia nigra pars reticulata (SNr) and the internal segment of the globus pallidus (GPi; entopeduncular nucleus in rodents). These neurons are referred to as direct pathway SPNs (dSPNs), because their axons project directly to the output nuclei, in addition to local striatal targets and the external segment of the globus pallidus (GPe, or simply GP in rodents). The second population of SPNs projected only to local striatal targets and the GPe and came to be known as indirect-pathway SPNs (iSPNs). Most dSPNs also contain the neuropeptides dynorphin and substance P, whereas most iSPNs contain the neuropeptide enkephalin (Beckstead and Kersey 1985).
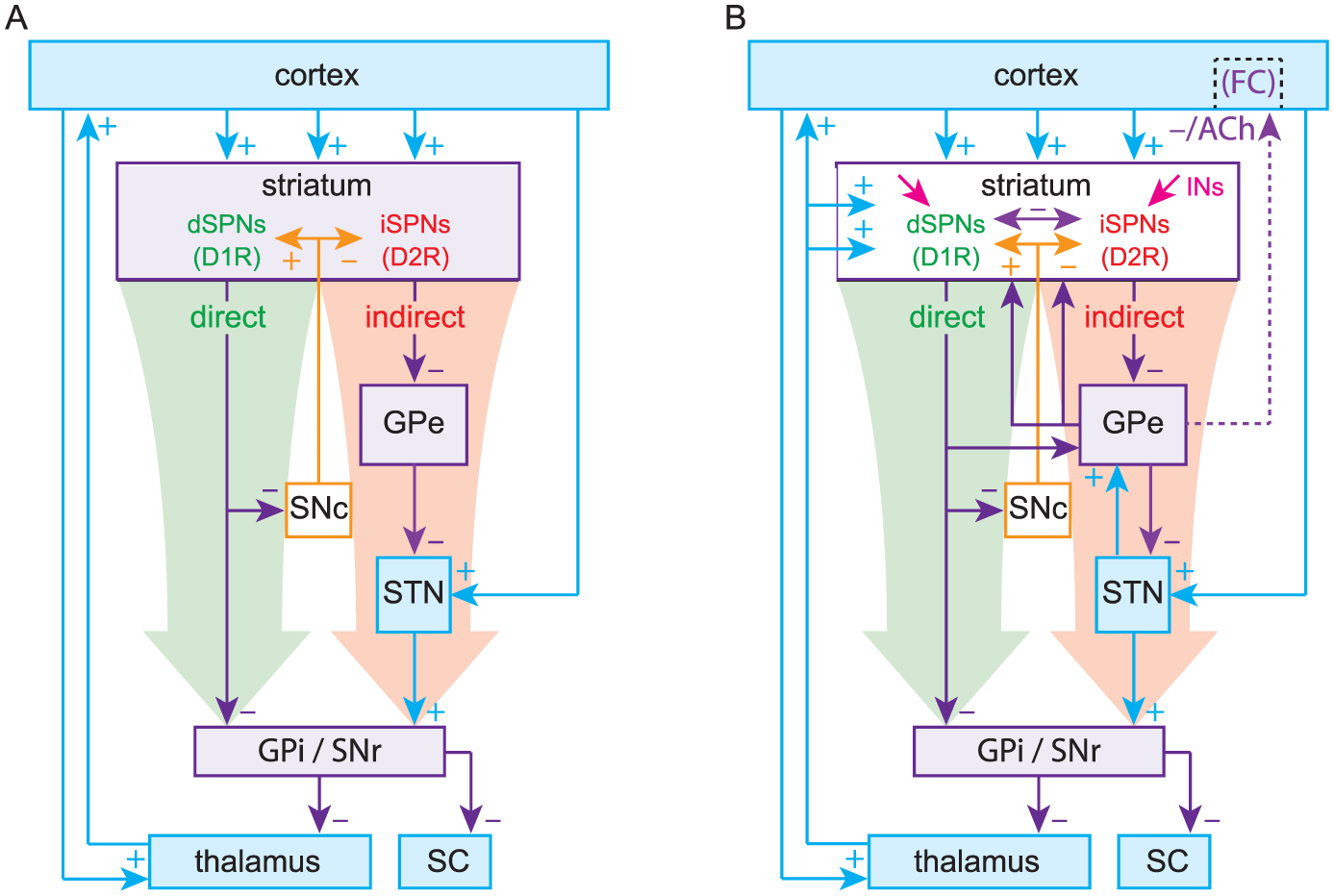
(A) The original Box-and-Arrow model, reformatted. (B) An updated model includes the thalamostriatal, subthalamopallidal (STN → GPe), pallidostriatal and pallidocortical pathways (Saunders and others 2015) as well as striatal interneurons and collaterals. d/iSPNs, direct/indirect pathway spiny projection neurons; GPe/i, external/internal segments of the globus pallidus; SNc/r, substantia nigra pars compacta/reticulata; SC, superior colliculus; INs, striatal interneurons; D1R/D2R, dopamine D1/D2 receptors; FC, frontal cortex; ACh, acetylcholine.
Another principle of BG function that was elaborated upon during this period is that of disinhibition—the process by which the target of an autonomously active population of inhibitory neurons is released from tonic inhibition when the inhibitory neurons are themselves inhibited (Chevalier and Deniau 1990). Work in non-human primates subsequently demonstrated that GABAergic SNr neurons tonically inhibit neurons in the superior colliculus (SC). Inhibition of SNr neurons by dSPNs disinhibits the SC, enabling the generation of saccades (Wurtz and Hikosaka 1986). Thus, a model of how the BG gates action began to emerge. Activation of the feedforward direct pathway, comprised of dSPNs inhibiting SNr/GPi neurons, disinhibits the targets of the BG (e.g., SC and thalamus) to enable movement. The direct pathway, thus, became known as the “GO” pathway of the BG.
At around the same time, subthalamic nucleus (STN) neurons were recognized to be glutamatergic and generate monosynaptic excitatory postsynaptic potentials (EPSPs) in SNr neurons (Kita and Kitai 1987; Nakanishi and others 1987). Because STN neurons receive GABAergic input from the GPe and project to the GPi/SNr, it was concluded that the indirect pathway (iSPNs to GPe to STN to GPi/SNr) forms a “dis-disinhibitory” (equivalent to an inhibitory) pathway, ultimately disinhibiting GPi/SNr output and suppressing movement. The indirect pathway came to be known as the “NO-GO” pathway of the BG.
The pinnacle of the formulation of the box-and-arrow model was the discovery that dSPNs and iSPNs can also be distinguished by their surface expression of dopamine receptors: dSPNs express D1 receptors (D1Rs), which generally enhance neuronal excitability, whereas iSPNs express D2 receptors (D2Rs), which generally reduce neuronal excitability (Gerfen and others 1990; Gerfen and Surmeier 2011). This distinction provided a straightforward explanation for the facilitating action of dopamine in the striatum. When dopamine is released in the striatum, it activates D1Rs on dSPNs to increase activation of the direct pathway and D2Rs on iSPNs to decrease activation of the indirect pathway, biasing the BG toward a “GO” state. By the same logic, it was argued that when dopamine is depleted, as in Parkinson’s disease (PD), the BG would be biased toward a perpetual “NO-GO” state, which could explain the hypokinetic nature of PD (Fig. 2). Using a similar argument (but independent of dopamine) the box-and-arrow model could also account for Huntington’s disease (HD), characterized by involuntary hyperkinetic choreic movements and a massive loss of SPNs. A preferential loss of iSPNs in early stages of HD would presumably weaken the indirect pathway and bias the network toward a perpetual “GO” (i.e., choreic) state, wherein the output nuclei of the BG are underactive (Fig. 3).
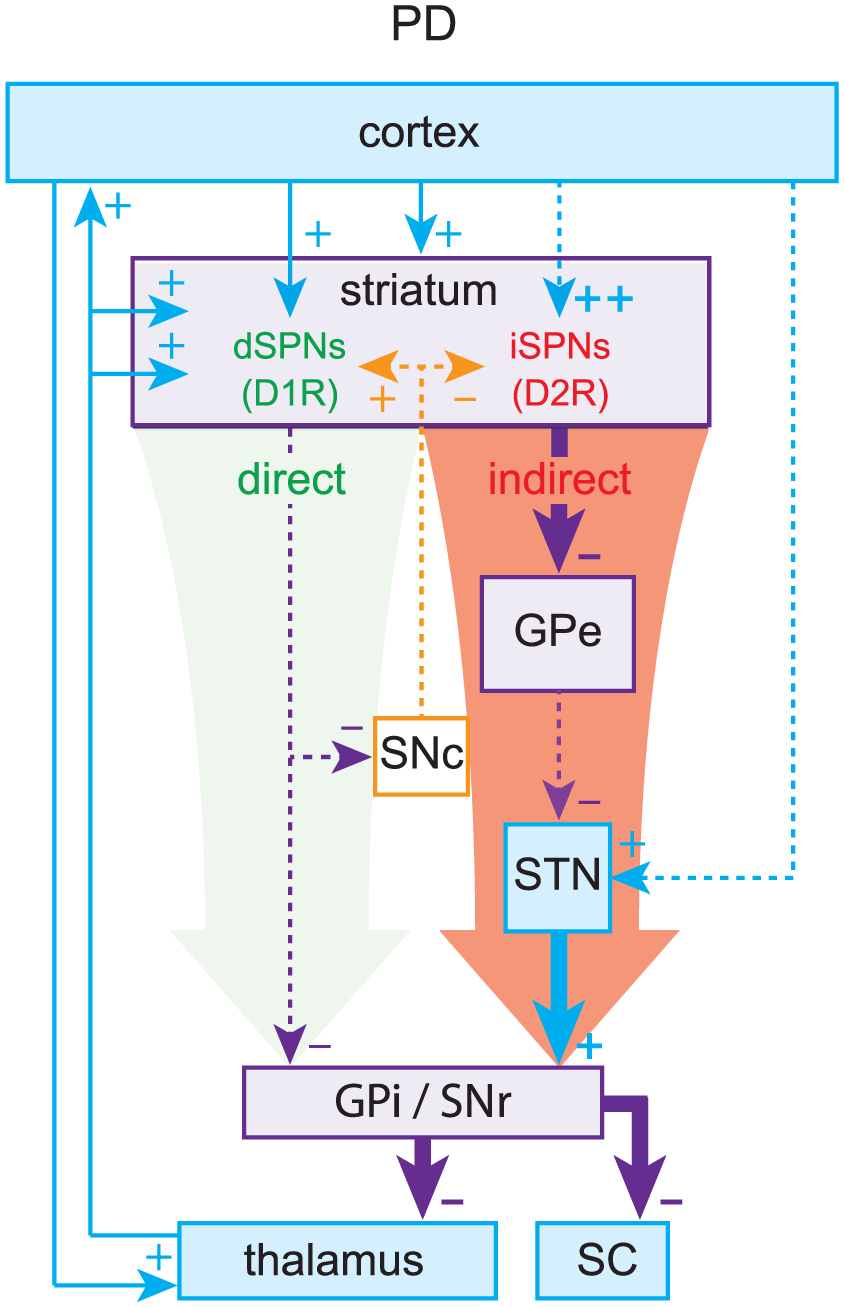
Parkinson’s disease (PD). Loss of dopamine weakens the direct pathway and strengthens the indirect pathway, leading to elevated inhibitory outflow from the basal ganglia (BG).
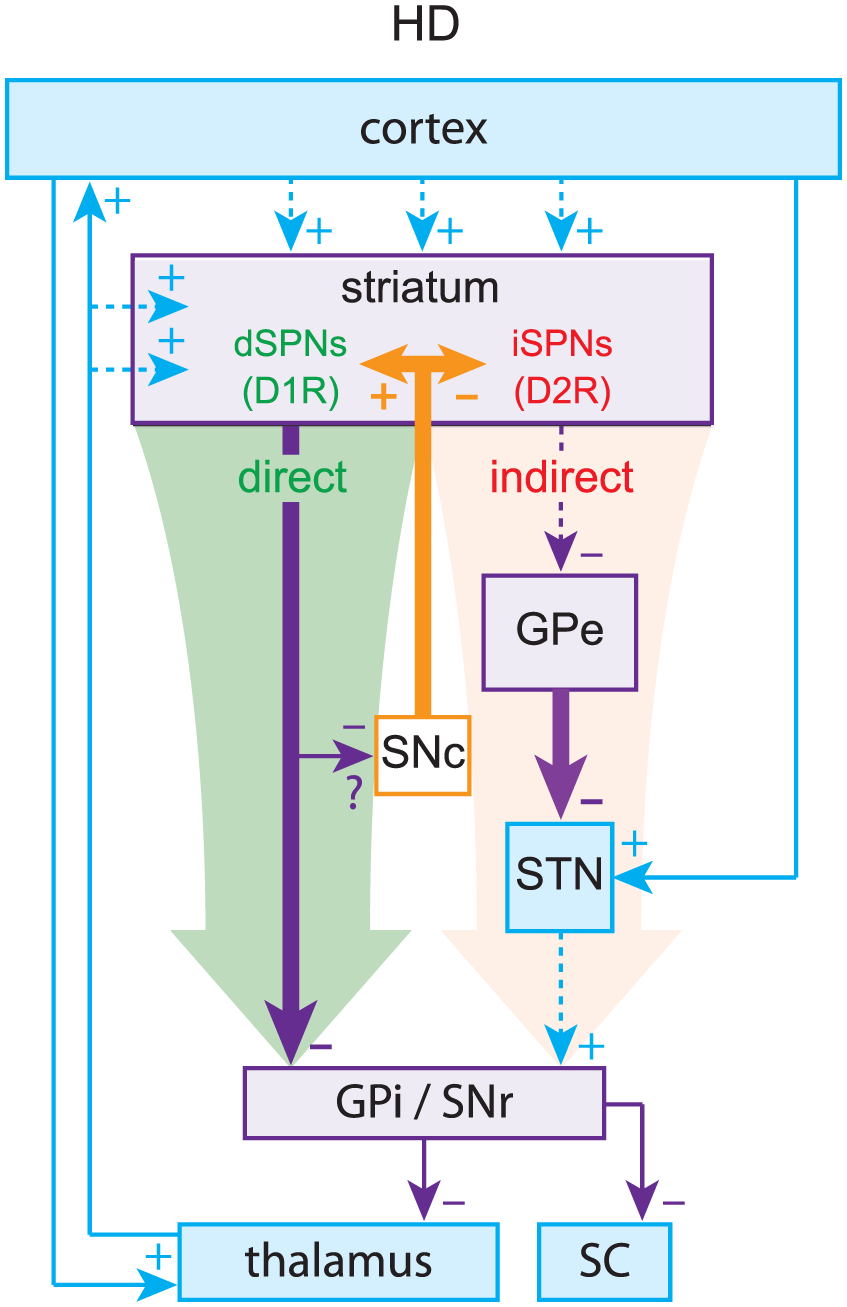
Huntington’s disease (HD). Preferential loss of indirect pathway spiny projection neurons (iSPNs) leads to relative overactivation of the direct pathway, leading to decreased inhibitory outflow from the basal ganglia (BG).
Thus, the strength of the box-and-arrow model was in its ability to explain changes in the basal firing rates of neurons within the various BG nuclei, which in turn presumably explains the hypokinetic and hyperkinetic motor symptoms of PD (e.g., akinesia/bradykinesia) and HD (e.g., hyperkinesia/chorea in early stages and hypokinesia/bradykinesia in late stages). Because tremors, which are a cardinal symptom of PD, are dynamic by nature, no attempt was made initially to use the box-and-arrow model to explain them. However, if the circuit or loop structure of the box-and-arrow model is taken into consideration, ideas from control theory can show that rhythmic activity can easily arise in these models as destabilization of a negative feedback loop, which could in turn explain tremorgenesis (see Box 1). Nevertheless, showing a causal relationship between pathological oscillations and tremor has proved tricky.
Oscillations and synchrony in the basal ganglia (BG): A dynamical extension of the box-and-arrow model.
At about the time of the formulation of the box-and-arrow model, studies began to show that neural activity in the cortico-BG circuits of human PD patients and animals with experimental parkinsonism exhibited abnormal oscillatory patterns of activity in the theta (5-8 Hz) and beta (15-40 Hz) ranges (Bergman and others 1994; Brazhnik and others 2014; Goldberg and others 2002; Goldberg and others 2004; Hutchison and others 1997; Hutchison and others 2004; Lenz and others 1988; Levy and others 2000; Mallet and others 2008; Nini and others 1995; Raz and others 1996). Because resting tremor is one of the cardinal symptoms of the disease, it was tacitly assumed that the oscillatory activity in the BG must cause the tremor, although this has never been demonstrated convincingly. Moreover, there is evidence that brain regions, such as the cerebellum, that are not part of the cortico-BG circuit, are implicated in the tremor as well (Wu and Hallett 2013). Nevertheless, the discovery of these oscillations sparked interest in the hypothesis that the known BG circuitry—as reflected in the box-and-arrow model—could in and of itself give rise to oscillations. This hypothesis was buttressed by a seminal article by Dietmar Plenz and Steve Kitai (Plenz and Kital 1999) that demonstrated the spontaneous generation of oscillatory activity in the STN and GP in the lower and sub-delta (0.5-2 Hz) range in an organotypic co-culture of cortex, striatum, STN and GP. Importantly, they demonstrated that it was the sub-circuit comprised of the reciprocally connected GP and STN in the culture that was necessary for the generation of oscillations (Fig. 1B). Note that the reciprocal connection between the GP and STN was entirely absent from the box-and-arrow model (Fig. 1A). We now know that the capacity of this sub-circuit to generate oscillations is related to the fact that GP and STN neurons are themselves autonomous pacemakers. Thus, their reciprocal excitatory (STN to GP) and inhibitory (GP to STN) inputs create something akin to a central pattern generator, whose ability to generate rhythmic output arises from a similar excitatory–inhibitory coupling of autonomously bursting neurons in the spinal cord (Bevan and others 2002; Pearson 1976; Terman and others 2002). Incidentally, because many neuronal types in the BG are autonomous pacemakers, including dopaminergic SNc neurons and some striatal interneurons (see Box 2), the study of mechanisms of robust pacemaking has become one of the central themes of BG research for both experimentalists and modelers (Atherton and others 2008; Chan and others 2004; Chan and others 2007; Gunay and others 2008; Surmeier and others 2005; Wilson and Bevan 2011), and has given us new insights into how channelopathies that affect pacemaking contribute to the pathophysiology of PD and other BG-related movement disorders (Atherton and others 2016; Chan and others 2007; Chan and others 2011; Wilson and Bevan 2011).
While the GP-STN circuit is widely thought to be the driver of abnormal oscillatory activity in the BG, recent work by Nicolas Mallet has shown that this cannot be the entire story. As mentioned above the appearance of beta band oscillations is a hallmark of PD and experimental parkinsonism. In a very elegant study, he used optogenetics to silence neural activity in various BG structures in hemi-parkinsonian mice in vivo (where dopaminergic neurons were destroyed chemically only in one hemisphere). He found that the beta band activity was abolished only when GP activity was silenced, but not when the STN was silenced (N. Mallet, personal communication), which argues against the GP-STN sub-circuit as the driver of parkinsonian beta oscillations.
An alternative hypothesis is that rhythmogenesis can be conceived as arising from a delayed negative-feedback loop composed of the entire cortico-BG circuit, with the delays arising from lags in polysynaptic transmission and integration. Control theory predicts that delayed feedback loops can become destabilized if the gain in the circuit is too high. A day-to-day analogy is the procedure of adjusting the heat of the hot-water tap for a shower. If the boiler is positioned far away from shower tap (hence the delay), it will take a few cycles of “too cold”/“too hot” to get it just right. The ability to change the gain of the adjustment (make smaller and smaller corrections with the tap) make it possible to reach a desired comfortable level with only a few cycles of over- and undershooting. Now, if the person in the shower is incapable of making small adjustments (i.e., her gain is too high) she could find herself incapable of reaching a stable fixed temperature but instead experiencing a never-ending oscillation between “too hot” and “too cold.”
A hypothesis originally proposed by one of us (Goldberg and others 1999) is that the imbalance between the direct and indirect pathways in PD results in an increased gain along the circuit which gives rise to oscillations (Leblois and others 2006). To see this, we consider the box-and-arrow circuit in the parkinsonian state. As described in the accompanying figure, the circuit can be reduced to a two-population network where “cortex” represents thalamocortical circuitry and where the entire multisynaptic pathway leading from the cortex to the GPi is collapsed to a single effectively excitatory link from “cortex” to GPi. The assumption is that the imbalance between the direct and indirect pathways that occurs in PD translates to an increase in the effectively positive gain along this pathway. Under these circumstances the “cortex” and GPi settle in an out-of-phase oscillation. The effective cortical excitation reaches the GPi with a delay (e.g., half a cycle late). The resulting excitation of GPi increases the inhibition returned to the “cortex”. This causes the previous cortical excitation to wane, which in turn reduces the excitation of the GPi. The GPi then disinhibits the “cortex”, and so the “cortex” excites the GPi again. This process repeats ad infinitum resulting in stable rhythmic oscillations.
Another dynamical property of BG activity that has become a hallmark of parkinsonism is the appearance of abnormal synchronous activity throughout the cortico-BG circuitry in both human and non-human primates (Goldberg and others 2002; Goldberg and others 2004; Nini and others 1995; Raz and others 1996). It is possible that abnormal synchrony, particularly in the output from motor cortex (Devergnas and others 2014; Goldberg and others 2002; Pasquereau and others 2016), ultimately co-activates antagonistic muscles and thus underlies akinesia and cogwheel muscle rigidity that are hallmarks of PD (Goldberg and others 2002). The appearance of abnormal synchrony in the GP is particularly remarkable, given that in healthy individuals the neural activity of GP neurons is found to be entirely decorrelated, despite the large convergence of inputs to the GP from the striatum and the STN. It is currently believed that collective GP activity is actively decorrelated (Goldberg and others 2013; Wilson 2013), with the purpose of optimizing information compression and transfer and the maintenance of distinct information channels that course through the GP. The loss of this active decorrelation is thought to contribute to the pathophysiology of PD. It has even been proposed that the therapeutic action of deep brain stimulation is achieved by restoring the decorrelated state and disrupting abnormal synchrony (Wilson and Bevan 2011).
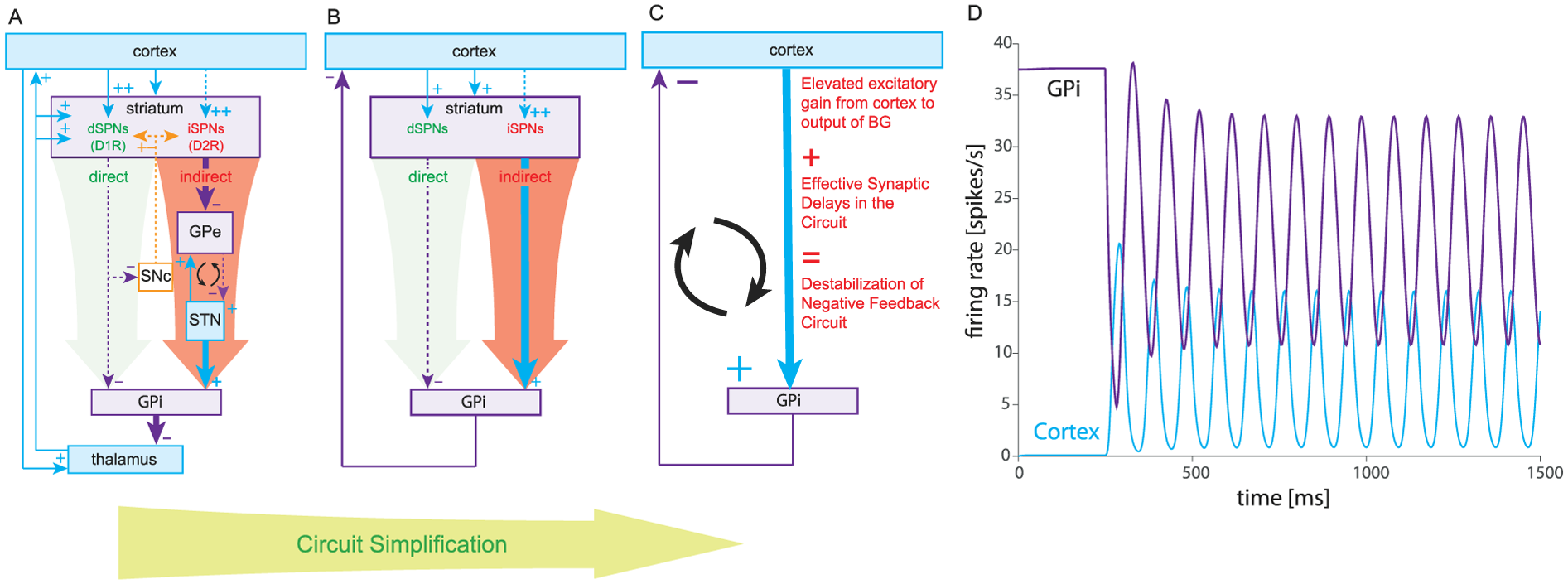
Cortico-basal ganglia oscillations in the box-and-arrow model of Parkinson’s disease (PD). (A) Box-and-arrow diagram for PD, including the recurrent connections between the subthalamic nucleus (STN) and external segment of the globus pallidus (GPe), which are considered a possible driver of parkinsonian oscillations (the weakened cortex-STN link (Chu and others 2017) is omitted). (B) Same network after subsuming the thalamus into the cortex (both are excitatory) and after subsuming the indirect pathway into an effective excitatory direct pathway that is enhanced in PD. (C) Further collapsing the direct and indirect pathway and the striatum as a whole into a delayed excitatory cortex-GPi (GPi is the internal segment of the globus pallidus) link whose gain is elevated relative to the normal condition. (D) Increasing the gain in a simplified basal ganglia (BG) network (at t = 250 ms) can give rise to oscillations due to synaptic delays along the entire cortico-BG circuit.
In summary, the conceptually simple, elegant, evidence-based box-and-arrow model provided a first theoretical framework to (a) explain the collective action of the BG in health and disease (Albin and others 1989; DeLong 1990), (b) generate testable experimental predictions (Bergman and others 1994; Boraud and others 1996; Cui and others 2013; Filion and Tremblay 1991; Kravitz and others 2010) and (c) be readily taught to the next generation of neuroscientists and medical practitioners. Most important, it helped establish functional neurosurgery as a justifiable method to normalize the mean activity of the various BG nuclei in disease states (Bergman and others 1990; Breit and others 2004; Limousin and others 1995). Indeed, deep brain stimulation of targeted BG nuclei has helped over a hundred thousand patients worldwide since the early 1990s (Ponce and Lozano 2010). For these reasons, the box-and-arrow model has become the prevailing dogma in the field, which adorns medical and neuroscience textbooks (Berne and others 2010; Squire 2013).
Over the course of the two and a half decades since the formulation of the box-and-arrow model, our knowledge of BG anatomy, biochemistry, circuitry, and physiology has expanded, giving us new insights into the pathogenesis of BG-related movement disorders and elucidating key predictive limitations of the original model. Although many transformative discoveries have been made throughout the field, our focus here will be on the striatum, in particular the dorsal striatum, which is associated with motor behavior. In particular, we focus on two areas in which there have been great strides in understanding the structure and function of the striatum: striatal microcircuitry and synaptic plasticity. In each case, we will reference various movement disorders (with an emphasis on PD and HD) to demonstrate how the current conceptualization of striatal pathophysiology is related to maladaptive changes in striatal circuit function.
The Striatum: The Heart of the Box-and-Arrow Model
The striatum can be considered the heart of the box-and-arrow model. Since it houses all dSPNs and iSPNs, it is the site of the initial divergence of the direct and indirect pathways and is where dopamine acts to facilitate action (Alexander and Crutcher 1990; Gerfen and others 1990; Gerfen and Surmeier 2011; Surmeier and others 2009; Tritsch and Sabatini 2012). It is also where dopamine depletion causes circuit changes thought to lead to the hypokinetic symptoms of PD and where the selective dysfunction and ultimate loss of iSPNs is thought to underlie early-stage HD (Albin and others 1989; DeLong 1990; Plotkin and Surmeier 2015; Reiner and others 1988). Both effects have been attributed to an “imbalance” between the direct and indirect pathways (see Figs. 2 and 3). Studies using optogenetic techniques (i.e., the selective expression of light activated ion channels in mammalian tissue and transgenic mouse lines in a cell-type specific manner) to selectively activate either the direct or the indirect pathway in freely moving mice have lent support to the overall antagonistic nature of these pathways both in health and disease (Kravitz and others 2010). However, such forced activation of striatal pathways is akin to restricting a pianist to strike two chords: one composed of all the white keys and the other of all the black keys. Such a manipulation would eradicate any finesse in the neural output of the striatum. Indeed, recent studies have clearly demonstrated that dSPNs and iSPNs are co-activated during normal motor behavior in freely moving mice and cannot be perceived as strictly antagonistic to each other (Cui and others 2013).
The arrows representing striatal output in the model are encoded exclusively by GABAergic SPNs, the activity of which is constrained and shaped by the complex microcircuity described in the boxes and text of this review (and much of which is not represented in the box-and-arrow model). There are many factors that determine the relative weights of the diagrammatic arrows in Figure 1A, including the biophysical properties of dSPNs and iSPNs themselves and the synaptic strength of their afferent inputs. The first thing that should be noted is that iSPNs are intrinsically more excitable than dSPNs, suggesting that there is an innate aptitude for imbalanced striatal output (Cepeda and others 2008; Gertler and others 2008; Kreitzer and Malenka 2007). Alterations in SPN intrinsic excitability are common in disorders involving the striatum. For example, one of the most robust physiological alterations in genetic mouse models of HD is an increase in SPN input resistance (Dvorzhak and others 2013; Klapstein and others 2001; Plotkin and Surmeier 2015). The net effect is to make SPNs more excitable. This increase in input resistance may predominantly occur in dSPNs (Raymond and others 2011), thus biasing striatal output toward the direct pathway, consistent with hyperlocomotion associated with chorea. Changes in intrinsic excitability also occur in PD models, which will be discussed below.
The second thing to note about SPN physiology is that both dSPNs and iSPNs have remarkably hyperpolarized resting membrane potentials, near −90mV, which is due to the inward rectifying potassium channel they express (Gerfen and Surmeier 2011; Shen and others 2007; Wilson and Kawaguchi 1996). What this means is that SPNs are quiescent at rest and require substantial excitatory synaptic drive to approach spike threshold and fire action potentials. Therefore, in the absence of afferent stimulation, the striatum can essentially be thought of as “silent,” imposing no inhibitory cues on the downstream nuclei of the basal ganglia. SPNs require coordinated, converging synaptic activation to overcome their hyperpolarized resting membrane potential and fire action potentials (Goldberg and others 2003; Stern and others 1998; Wilson and Kawaguchi 1996).
How does converging excitatory input drive SPN output? Early studies in rodents observed that bouts of converging synaptic activation likely originating from the cortex can depolarize SPNs from their silent hyperpolarized resting membrane potential (referred to as the “down state”) to a depolarized “up state” from which action potentials can fire. While these states were first discovered in non-anesthetized animals, the particular anesthesia influences their shape and duration (Goldberg and others 2003; Mahon and others 2001; Stern and others 1998; Wilson and Groves 1981; Wilson and Kawaguchi 1996). Advances in optical tools led to the discovery that these “state transitions” can be induced by activating only about a dozen glutatmatergic inputs to distal SPN dendrites (where they locally induce prolonged plateau potentials that can outlast the synaptic stimuli by hundreds of milliseconds), provided that they are convergent in time and space (Du and others 2017; Plotkin and others 2011). Reliance on so few synapses (compared to original estimates of hundreds or more) greatly increases the computational capacity of SPNs (Blackwell and others 2003; Plotkin and others 2011) and suggests that the generation of dendritic plateaus may represent a mechanism by which meaningful synaptic associations can gate SPN output. What this means is that cortical activation of striatal output is not necessarily a linear process, as implied by the box-and-arrow model, and offers a clue as to how the striatum can begin to filter out cortical inputs that are potentially less relevant.
Striatal Afferents and Microcircuitry
Both dSPNs and iSPNs receive highly convergent glutamatergic afferents from numerous cortical and subcortical regions. There is some evidence for preferential innervation of pathways, for example functional responses to cortical pyramidal tract neuron afferents are larger in dSPNs (Kress and others 2013; but see Deng and others 2014; Lei and others 2004; Reiner and others 2010) and afferents originating from motor versus limbic-associated regions form biased connections on iSPNs and dSPNs, respectively (Wall and others 2013), but to date no inputs have been discovered that are SPN-type specific. This points out an important aspect of the model—though the direct and indirect pathways are depicted as discrete entities, there is tremendous overlap in their inputs and engagement.
Corticostriatal connections are roughly organized in a somatotopic fashion, with a consequence being that the striatum may connect diverse cortical regions to the thalamus via anatomically and functionally parallel (but we now know not fully independent) loops (Haber 2003; Parent and Hazrati 1995). Unlike structures such as the cortex and hippocampus, the striatum is not laminarly organized. However, postmortem histological analyses (such as acetylcholinesterase and mu opioid receptor expression) revealed that the striatum has a compartmental organization. About 10% of the striatum can be classified as striosomes (or patches), which are surrounded by the more prominent matrix compartment (Gerfen 1984; Graybiel and Ragsdale 1978). Functionally, we are just starting to understand the full significance of such compartmentalization, but studies suggest that SPNs residing within striosomes mediate the establishment of psychostimulant-induced motor stereotypies and guide cost-benefit decision-making (Canales and Graybiel 2000; Friedman and others 2017). Both afferents to and efferents from each compartment differ, with striosomes receiving more frontal cortex and limbic-associated inputs and directly innervating dopaminergic neurons in the substantia nigra pars compacta (Crittenden and others 2016; Fujiyama and others 2011), though the degree of this segregation has recently been questioned (Smith and others 2016). Although not explicitly diagrammed in the classic box-and-arrow model, it is a conceptually straightforward addition. It is an important addition as well, as compartment-specific pathology occurs in both HD and PD (Crittenden and Graybiel 2011; Hedreen and Folstein 1995).
By far the most prominent afferents to dorsal striatum SPNs are from the cortex and thalamus (in particular the centromedian (CM) and parafascicular (Pf) nuclei (Doig and others 2010; Dube and others 1988; Kemp and Powell 1971; Wilson and others 1990); though thalamic inputs were omitted from the original box-and-arrow model (Albin and others 1989; DeLong 1990), they have since become a standard addition (Alexander and Crutcher 1990; Squire 2013). Dysfunction of corticostriatal and thalamostriatal synaptic signaling is a hallmark of symptom progression in Huntington’s disease, with dysregulation of glutamatergic synapses within the striatum occurring well before frank striatal and cortical cell death (Deng and others 2013; Plotkin and Surmeier 2015; Raymond and others 2011; Zuccato and others 2010). These changes are complex, but overall there is a gradual synaptic disconnect between SPNs and their cortical inputs (Cepeda and others 2003; Raymond and others 2011). Just as cell death is first observed in iSPNs and later dSPNs (Reiner and others 1988), progressive SPN-type specific alterations in corticostriatal synaptic transmission occur as well, albeit in a complex manner (Andre and others 2011; Plotkin and others 2014). Overall such circuit changes are consistent with the early hyperkinetic symptoms of HD (Albin and others 1989; Andre and others 2011; Plotkin and others 2014; Zuccato and others 2010). Alterations in corticostriatal inputs are observed in PD models as well: dopamine depletion reduces the density of axospinous synapses formed by the cortex on iSPNs, while leaving those on dSPNs intact (Day and others 2006). While this may at first seem counterintuitive in light of PD being a hypokinetic disorder, this spine loss likely reflects a compensatory attempt to counteract an acute increase in neuronal excitability resulting from diminished activation of D2Rs (Day and others 2006; Fieblinger and others 2014). This illustrates an important caveat of the classic box-and-arrow model: Relative changes in the activity of the direct and indirect pathways may reflect more than just a change in the balance of dSPN and iSPN activity (thickness of the arrows in the model), but a pathological bias in the cortical and subcortical network information that is being processed (Fieblinger and others 2014).
While it is obvious that SPNs are major players in striatal circuit function and pathophysiology, there are also other types of striatal neurons, all of which are interneurons (see Box 2). Among these are cholinergic interneurons (CINs), which had long been implicated in movement disorders, but like all other interneurons are not clearly integrated into the model (Barbeau 1962; Lehmann and Langer 1983) (Fig. 1B). Anticholinergic therapy (administration of muscarinic receptor blockers) was widely used to treat striatum-associated disorders such as dystonia and PD before the advent of dopamine replacement therapy (Fahn 1983; Jankovic 2013; Lang and Blair 1989; Pisani and others 2007). Conversely, brain-penetrating acetylcholinesterase inhibitors were shown to reduce chorea in HD patients (Aquilonius and Sjostrom 1971). These clinical findings, in addition to experimental evidence that showed changes in the expression of dopaminergic and cholinergic markers in the striatum were inversely correlated in disease states, led to the famous striatal dopamine–acetylcholine imbalance hypothesis of BG-related movement disorders (Barbeau 1962; DeBoer and others 1996; Ding and others 2006; Lehmann and Langer 1983). CINs are the source of striatal acetylcholine (ACh) tone and must therefore be associated with the therapeutic effect of anticholinergics, but they are nevertheless glossed over in the model. This is a crucial issue to rectify, as it is now appreciated that cholinergic signaling (primarily through muscarinic receptors) mediated by CINs plays a profound role in modulating SPN excitability and synaptic transmission (Calabresi and others 1999; Deffains and Bergman 2015; Day and others 2008; Goldberg and others 2012; Pakhotin and Bracci 2007). As in the case of SPNs, alterations in cortico- and thalamo-striatal inputs to CINs are observed in movement disorders such as PD and HD (Aceves Buendia and others 2017; Holley and others 2015; Tanimura and others 2016). Whether these are homeostatic responses that aim to restore the dopamine–acetylcholine balance or maladaptive responses that exacerbate the imbalance is unclear. Furthermore, CINs have recently been shown to directly promote local dopamine release from SNc axonal terminals in the striatum (via a nicotinic acetylcholine receptor–mediated mechanism) (Threlfell and others 2012), highlighting the oversimplification and inherent caveats of the dopamine-acetylcholine imbalance hypothesis of striatal movement disorders.
Menagerie of striatal interneurons and GABAergic afferents.
In rodents, SPNs constitute more than 95% of striatal neurons. The remaining 2% to 4% of striatal neurons are composed of a single type of cholinergic interneuron (0.5%-1.5%) (see figure panel B) and several types of GABAergic interneurons (approximately 2%) (see figure panel C) (Assous and Tepper 2018; Graveland and DiFiglia 1985; Munoz-Manchado and others 2016). These interneurons exert powerful influence over SPN dynamics both on a moment-by-moment time scale by activating pre- and postsynaptic receptors on SPNs and on a slower time scale by influencing the intrinsic excitability of SPNs and synaptic plasticity. Many of these interneurons are also targeted by dopamine fibers, as well as other intrinsic and extrinsic inputs (Assous and Tepper 2018).
Cholinergic interneurons (CINs). CINs are aspiny neurons with large (20-50 µm long) somata and overlapping axonal and dendritic arbors that span about a third of the striatum. CINs are autonomous pacemakers (Bennett and Wilson 1999), meaning that they possess intrinsic membrane currents that compel them to constantly depolarize to action potential threshold independently of their synaptic inputs (Bennett and others 2000; Goldberg and Wilson 2010; Maurice and others 2004). This pacemaking action functions to maintain a perpetual ACh tone in the striatum. Because the axonal arbor of CINs is so extensive and space-filling, with release sites 1 µm apart, this perpetual tone is maintained throughout the whole volume of the striatum (Chang and Kitai 1982; DiFiglia and Carey 1986; Goldberg and Wilson 2010). ACh release is further modulated by direct glutamatergic and dopaminergic inputs that regulate CIN activity (see figure panel B).
CINs are major modulators of SPN intrinsic excitability, as well as synaptic transmission and synaptic plasticity of SPN afferents. This is mediated by muscarinic ACh receptors (mAChRs), which can be found on axon terminals and all striatal types (Goldberg and others 2012). Activation of presynaptic M2 type mAChRs on cortical axon terminals curbs cortico-striatal synaptic transmission (Higley and others 2009; Pakhotin and Bracci 2007), whereas postsynaptic M1 mAChRs amplify transmission by momentarily increasing SPN excitability (Goldberg and others 2012).
ACh release in the striatum also activates nicotinic ACh receptors (nAChRs) located on various GABAergic interneurons. This mediates di-synaptic feedforward inhibition onto SPNs as well as di-synaptic feedback inhibition onto other CINs (English and others 2012; Sullivan and others 2008; Witten and others 2010). Furthermore, activation of presynaptic nAChRs on dopaminergic nigrostriatal axon terminals can directly induce dopamine release, independent of somatic activity (Threlfell and others 2012). Adding a final level of complexity, it is now known that dopamine fibers themselves can co-release GABA (Tritsch and others 2012), raising the possibility that fast di-synaptic inhibition from CINs to SPNs can be mediated by dopaminergic fibers, as well.
Fast spiking interneurons (FSIs). The first and most widely studied GABAergic interneuron is the parvalbumin-positive (PV+) FSI that mediates strong feedforward cortical inhibition of SPNs (English and others 2012; Kawaguchi 1993). FSIs are hyperpolarized, have narrow action potentials and can fire at high rates often in a stuttering fashion (Kawaguchi 1993). These cells are one of the few examples of neurons in the mammalian CNS that exhibit class II Hodgkin (1948) excitability, meaning that when they transition from quiescence to firing they discharge at a finite minimal firing beneath which they cannot fire (Assous and Tepper 2018). FSIs are interconnected by gap junctions (Kita and others 1990), similar to PV+ basket cells in the cortex, and can form a syncytium that synchronizes their inhibition throughout the striatum. FSIs also receive direct dopaminergic innervation (see figure panel C). It should be noted that despite initial conjectures, FSIs do not mediate the di-synaptic inhibition of SPNs by CINs.
Plateau low-threshold spike interneurons (PLTSIs). Another GABAergic interneuron that mediates cortical feedforward inhibition is the neuropeptide-Y–positive/somatostatin-positive/nitric oxide synthase–positive (NPY+/SST+/NOS+) PLTSI. Its name summarizes its unique discharge properties in response to current injections and synaptic stimulation. Depolarizing current injections evoke a plateau potential and action potentials; depolarizing from a sufficiently hyperpolarized potential elicits a calcium-mediated low-threshold spike (LTS) that can also trigger sodium spikes (Kawaguchi 1993). PLTSIs are also autonomous pacemakers (Assous and Tepper 2018; Beatty and others 2012), and exhibit both single spiking and bursting discharge. It is likely that tonic activity is necessary for maintaining levels of the various neuromodulators and neuropeptides that PLTSIs release, which may be determined by the firing pattern (Beatty and others 2012). The axonal arbors of PLTSIs are unbranched and sparse but can span the entire extent of the striatum. PLTSIs do not receive input from CINs and only about 20% receive inputs from FSI (Szydlowski and others 2013). They do, however, receive dopaminergic inputs (see figure panel C).
Calretinin-positive (CR+) interneurons. CR+ interneurons were among the first GABAergic interneurons to be discovered, and in primates are composed of several subtypes that together outnumber FSIs and PLTSIs. Nevertheless, very little is known about them or their connectivity within the microcircuit, and only recently have articles begun to appear characterizing their activity in vivo (Garas and others 2018) (see figure panel C).
Tyrosine hydroxylase–positive (TH+) interneurons. In recent years, two new classes of GABAergic interneurons that receive both dopaminergic and cholinergic inputs have been rediscovered. TH+ interneurons are autonomously active, targeting PLTSIs and mediating cortical feedforward inhibition of SPNs (Xenias and others 2015). One type of TH+ interneurons is currently unique in that it is the only type of GABAergic interneuron that is known to receive synaptic inhibition directly from SPNs (Ibanez-Sandoval and others 2010).
Neuropeptide-Y–neurogliaform (NPY-NGF) interneurons. Another class of recently described GABAergic neurons are NPY+ interneurons (sometimes referred to as neurogliaform interneurons) (Ibanez-Sandoval and others 2011). NPY-NGF interneurons do not express SST or NOS, nor do they display any of the physiological properties of the PLTSIs. Instead, they physiologically resemble SPNs: hyperpolarized at rest and require significant depolarization to fire. NPY-NGF interneurons have a dense axon that innervates a radius of approximately 0.5 mm and are reciprocally coupled with CINs. Accordingly, NPY-NGF neurons were found to mediate di-synaptic feedforward inhibition of SPNs by CINs, di-synaptic feedback inhibition among CINs (English and others 2012; Sullivan and others 2008) and even di-synaptic feed-forward disinhibition of SPNs by FSIs (Lee and others 2017). NPY-NGFs are coupled by gap junctions, which presumably helps to synchronize and amplify their inhibitory influence (see figure panel C). NPY-NGF interneurons, along with SPN axon collaterals, have been proposed to be uniquely positioned to modulate SPN dendritic plateau potentials and resulting spiking (Du and others 2017).
Other interneurons. Our understanding of the complexity and heterogeneity of striatal interneurons is a work in progress, with new interneuron classes and connections still being discovered. For example, a recent study described a new GABAergic interneuron that only targets other interneurons, therefore affecting SPNs only by di-synaptic disinhibition, a novel concept in striatal microcircuitry (Assous and others 2018).
Extrinsic inputs to the striatal microcircuitry. The striatum receives extensive afferents from the cortex and SNc, but two important extrastriatal sources are not explicitly present in the original box-and-arrow model: the thalamus and globus pallidus (see figure panel D). The thalamus (e.g., CM and Pf) is an easy fix, and is the source of about 50% of the glutamatergic synapses onto SPNs (Dube and others 1988; Kemp and Powell 1971). Pf inputs also impinge on CINs, FSIs, NPY-NGF interneurons and TH+ interneurons. PLTSIs are an exception in that they do not receive direct excitatory input from the Pf but receive powerful di-synaptic inhibition via TH+ interneurons (Assous and others 2017). The biophysics of synaptic transmission and plasticity are often different between cortico- and thalamo-striatal synapses and the direction of this difference depends on the identity of the postsynaptic neurons being targeted (Aceves Buendia and others 2017; Ding and others 2008; Ding and others 2010).
While the existence of a pallidostriatal projection targeting interneurons has been known for decades (Bevan and others 1998; Goldberg and others 2003), recent work has identified a subclass of pallidal neurons (termed “arkypallidal” neurons) that project back to SPNs, CINs and some of the GABAergic interneurons. These pallidostriatal neurons are molecularly and physiologically distinct from the majority of “prototypical” pallidal neurons that project to the STN and output nuclei of the BG (Gittis and others 2014; Glajch and others 2016; Hegeman and others 2016; Mallet and others 2012).
Upon reflection on the current snapshot of striatal microcircuitry and its inputs and outputs (see figure) it seems at first sight overly complex. We are beginning to understand some of the rules of connectivity, but one thing is clear: reducing the whole striatal circuitry to a single box represented only by SPNs is no longer tenable.
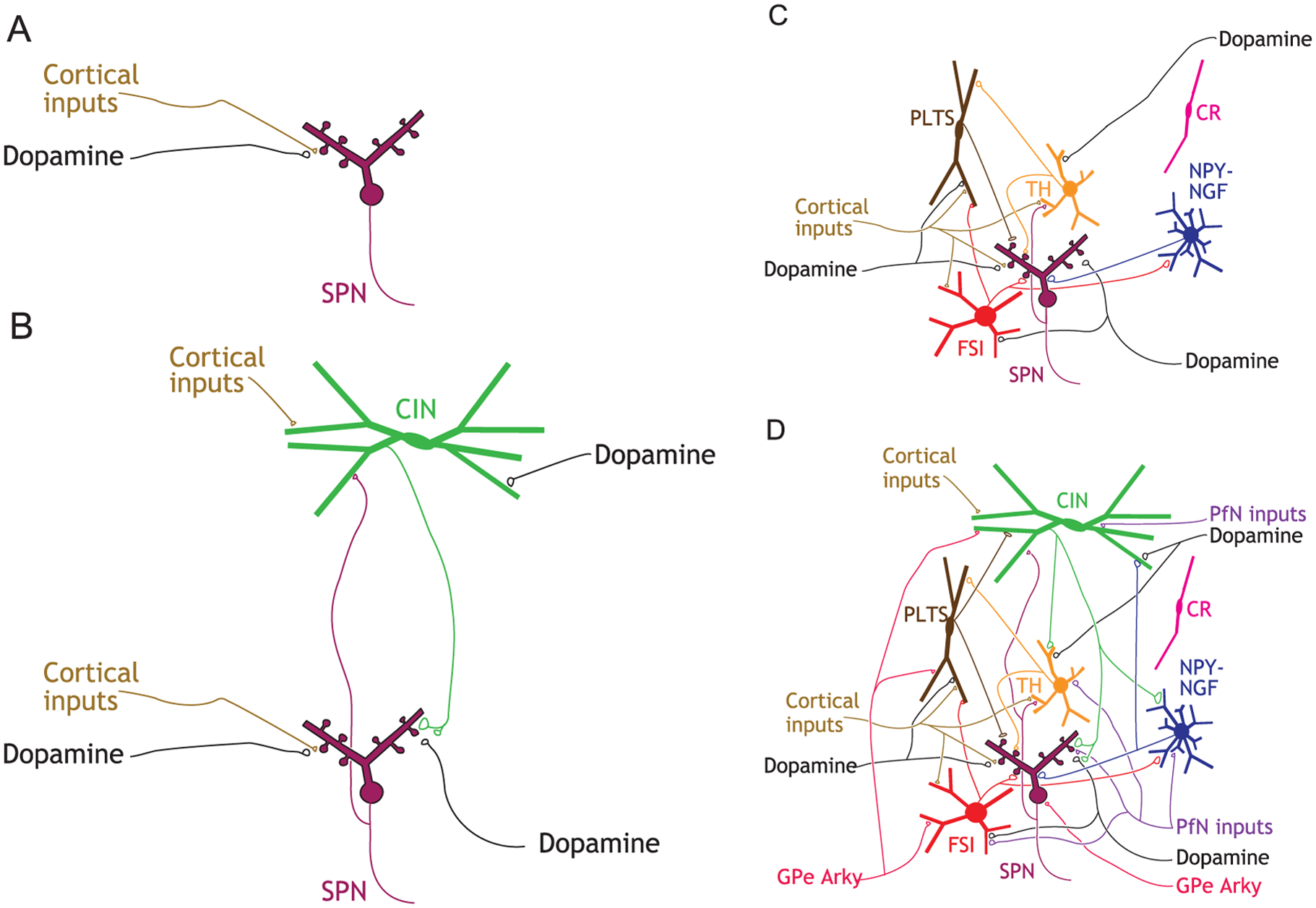
Striatal microcircuitry. (A) rendition of the striatum in original box-and-arrow model (with dSPN and iSPN collapsed to a single SPN), which includes only cortical and dopaminergic afferent inputs. (B) Cholinergic interneurons (CINs) are reciprocally (Chuhma and others 2011) connected to SPNs and terminate on dopamine axons. (C) GABAergic interneurons with their known connections to SPNs, reciprocal connections, and their cortical and dopaminergic inputs. (D) A current snapshot of known striatal circuitry, including afferent inputs from the GPe and the thalamic parafascicular nucleus (PfN). Highlights: (1) CINs and PLTSIs are tonically active autonomous pacemaker neurons; (2) CINs and TH interneurons are the only interneurons known to receive direct SPN input; (3) CINs and FSIs do not communicate with each other; 4) cortical and thalamic feedforward inhibition is mediated by a different subset of GABAergic interneurons. Abbreviations: see text.
The remaining 2% of striatal neurons (in rodents, in primates the percentages are considerably higher) are composed of multiple types of GABAergic interneurons, several of which co-release other signaling molecules (see Box 2) (Assous and Tepper 2018; Graveland and DiFiglia 1985; Kawaguchi and others 1995; Munoz-Manchado and others 2016; Tepper and Koos 2017). Because SPNs receive cortical innervation directly and because they (and only they) project outside the striatum, the first-approximation of the box-and-arrow model that neglects GABAergic interneurons seems reasonable (Fig. 1A). However, we now know that these interneurons exert a powerful influence over SPN dynamics and synaptic inputs both on a moment-by-moment basis through fast synaptic transmission and on a slower time scale by influencing the intrinsic excitability of SPNs and synaptic plasticity (Assous and Tepper 2018; Gittis and Kreitzer 2012; Koos and Tepper 1999; Logie and others 2013; Luo and others 2013; Owen and others 2018; Paille and others 2013; Rafalovich and others 2015; West and Grace 2004). Many interneurons are also targeted by dopamine fibers (Assous and Tepper 2018), endowing them with the ability to shape how dopamine release modulates striatal output. The advent of optogenetics has led to a recent explosion in our understanding of the rich connectivity and functional consequences of local striatal microcircuitry (see Box 2).
While pathological cholinergic signaling has been implicated in PD for decades (Barbeau 1962; Lehmann and Langer 1983), the idea that dysfunction of other interneurons fundamentally contributes to disease states has only recently garnered significant support. For example, although striatal GABAergic interneurons were once thought to be relatively spared from pathology in HD (Mitchell and others 1999), it is now known that the number of fast spiking interneurons (FSIs) is reduced in the striatum of symptomatic patients (Reiner and others 2013). Furthermore, technological advances in both the refinement of mouse models and experimental techniques have discovered that the firing rate of “plateau low-threshold spike” interneurons (PLTSIs) and SPN responses to FSIs increase in symptomatic HD mice, consistent with overall observations of elevated intrastriatal GABAergic synaptic transmission (Cepeda and others 2013). Pathological alterations in connectivity have also been observed. FSIs rapidly increase their connectivity to iSPNs (not dSPNs) in the 6-OHDA mouse model of PD, a circuit alteration that has a net effect of enhancing synchrony among iSPNs (Gittis and others 2011), and a phenomenon not readily predicted by the box-and-arrow model. Interneuron pathology occurs in other striatum-related movement disorders as well, such as Tourette Syndrome where there is a reduction in the number of FSIs and CINs (Kalanithi and others 2005; Kataoka and others 2010), and a hamster model of dystonia where there is a reduction in the number of several types of GABAergic interneurons (Gernert and others 2000; Sander and others 2006). Thus, the role of the striatum in movement disorders can no longer be fully appreciated without knowledge of the contribution of interneurons (see Box 2).
Though interneurons undoubtedly account for a major component of intrastriatal inhibition, they are not the only source—SPNs also form collateral inhibitory connections with other SPNs. Importantly such inhibition occurs within and between direct and indirect pathway SPNs (Cepeda and others 2013; Guzman and others 2003; Taverna and others 2008). Under normal conditions this inhibition is biased, with the indirect pathway exerting a stronger inhibition over its counterpart than vice-versa (Taverna and others 2008). Alterations in such collateral inhibition occur in mouse models of both PD and HD (Cepeda and others 2013; Taverna and others 2008). Thus, pathway-specific striatal output is not as simple as dSPNs or iSPNs being activated—ongoing activity of SPNs shapes not only the output of their own pathway but that of their “opposing” pathway as well, a phenomenon not predicted by the box-and-arrow model and a concept that must be taken into account when referencing “balance” between the two pathways.
Synaptic Plasticity and Circuit Adaptation
Perhaps the hardest thing to capture in a box-and-arrow diagram is the fact that neither the function of the boxes nor the strengths of the arrows are static entities. The dorsal striatum has been shown to be a key player in goal directed and habit learning (Gremel and others 2016; Shan and others 2014; Shan and others 2015; Smith and Graybiel 2016; Yin and others 2004), the substrate of which is thought to be the ability to change the weights of physiologically relevant synapses in a way that will optimize behavioral outcomes. Both dSPNs and iSPNs have the capacity to bi-directionally change the weights of their synaptic inputs through long-term potentiation (LTP) and long-term depression (LTD), though the road to get to this conclusion was arduous (Calabresi and others 1992a; Calabresi and others 1992b; Kreitzer and Malenka 2007; Lovinger 2010; Shen and others 2008; Surmeier and others 2009). It is also not immediately obvious how the model can account for the development of symptoms and side effects over the course of disease treatment. For example, prolonged administration of levodopa or
Although LTP and LTD occur at the same types of striatal synapses, they are not expressed at the same loci within a synapse. LTP ultimately requires the insertion of AMPA receptors into the postsynaptic membrane, while LTD requires postsynaptic production of endocannabinoids that then retrogradely act on presynaptic CB1 receptors to reduce neurotransmitter release probability (Calabresi and others 2007; Kreitzer and Malenka 2008; Lovinger 2010; Surmeier and others 2009) (see Box 3). This distinction is important, as it sets the groundwork for therapeutic target identification and design. Though the locus of expression differs, both LTP and LTD require combined pre- and postsynaptic activity, the timing of which can determine the sign of synaptic change (Fino and others 2005; Jedrzejewska-Szmek and others 2017; Paille and others 2013; Pawlak and Kerr 2008; Shen and others 2008). Whether this timing is Hebbian (LTP is induced by presynaptic followed by postsynaptic activity and LTD is induced by the opposite ordering) or anti-Hebbian has traditionally been a controversial topic (Fino and others 2005; Pawlak and Kerr 2008; Plotkin and others 2013; Shen and others 2008; Shindou and others 2011), but the reasons for this have begun to surface. Basal levels and timed transient elevations of dopamine, engagement of local GABAergic microcircuitry, the degree of cytosolic calcium elevation achieved at the synapse by paired pre- and post- synaptic activity, repetition of stimulation, SPN-type, and even the rapid conversion of LTD to LTP when particular circuit-level conditions are met all determine the sign of plasticity that is achieved by paired pre- and postsynaptic activity (Paille and others 2013; Plotkin and others 2013; Shen and others 2008; Shindou and others 2011; Shindou and others 2018; Yagishita and others 2014). Though the details of how each of these parameters contributes to spike timing dependent plasticity are beyond the scope of this review, we note these complexities to illustrate the myriad factors that determine the “weight” of each arrow in the box-and-arrow model, particularly under pathological conditions.
Striatal plasticity.
Corticostriatal LTP and LTD each involve their own sets of “key players.” For LTP, these include engagement of N-methyl-
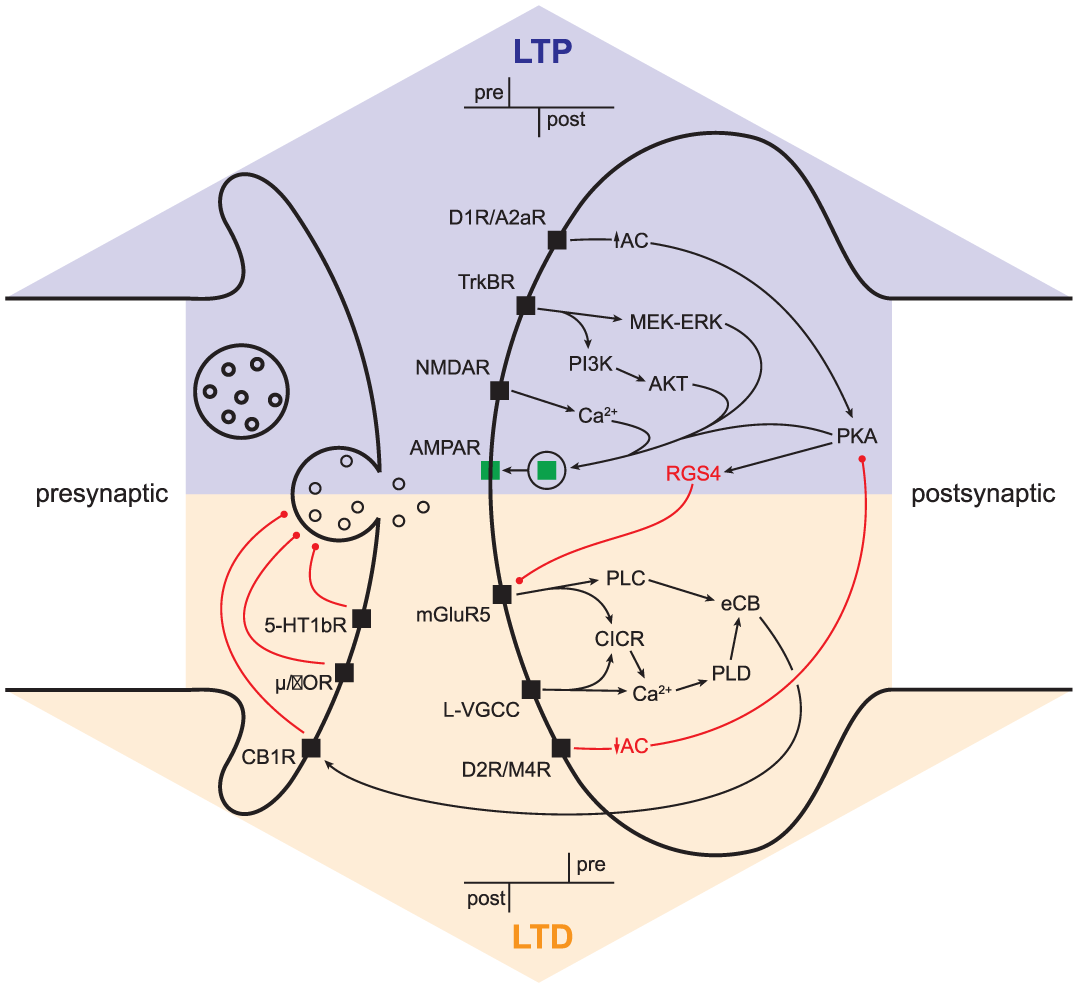
Synaptic plasticity. Pre- and postsynaptic signaling cascades responsible for long-term potentiation/depression (LTP/LTD). Abbreviations: MEK-ERK, mitogen-activated protein kinase kinase (MEK)–extracellular signal-regulated kinase (ERK); AKT, protein kinase B; PLC, phospholipase C; PI3K, phosphoinositide 3-kinase; CICR, calcium-induced calcium release; PLD, phospholipase D; eCB, endocannabinoid; µ/δOR, µ/δ opioid receptor; 5-HT1bR, 5-HT type 1b receptor; other abbreviations as in text.
While both dSPNs and iSPNs support LTP and LTD, the neuromodulators associated with the induction of each differs between the two neuron populations (see Box 3). The reason is readily apparent in the box-and-arrow model: D1Rs (which promote LTP) are only expressed in dSPNs, while D2Rs (which promote LTD) are only expressed in iSPNs (Gerfen and others 1990; Gerfen and Surmeier 2011). This leads to a conceptual problem: how do both dSPNs and iSPNs display bidirectional plasticity if D1 and D2 receptors are segregated in the two neuron populations? Groundbreaking work by the Surmeier group solved the mystery of how this can happen. Dopamine receptors are not the only neuromodulator receptors that show SPN-type subunit specificity. Though iSPNs do not express D1Rs, they do express A2a-type adenosine receptors, which engage the same Golf-linked G-protein coupled receptor (GPCR) signaling cascade and promote LTP in the same way D1Rs do in dSPNs (Shen and others 2008). Similarly, while dSPNs do not express D2Rs they do express M4-type muscarinic receptors (M4Rs), which feed into the same Gi-linked GPCR signaling cascade and promote LTD in the same way D2Rs do in iSPNs (Shen and others 2015). Though this endows both SPN populations with the ability to support bidirectional plasticity, two things should be noted: (1) Only one form of plasticity in each SPN type may directly reflect stored information that was encoded by behaviorally relevant elevations in dopamine and (2) disease-related impairments in dopamine release will have opposite effects on the sign of plasticity in the direct and indirect pathways. Indeed, the latter point was demonstrated by Shen and others (2008), who showed that dopamine depletion caused paired spike patterns that normally induce LTP to yield LTD in dSPNs and paired spike patterns that normally induce LTD to yield LTP in iSPNs. This phenomenon is consistent with the box-and-arrow diagram used to explain PD: weakening of the direct pathway and strengthening of the indirect pathway. But the way in which this happens differs- in the model pathway-specific output is globally elevated or reduced, while the above finding suggests that such changes may have a synapse-specific underpinning. This is an important point, as not all glutamatergic inputs to the striatum support the same forms of LTP and LTD (Plotkin and others 2014; Wu and others 2015), adding a layer of network-specificity not afforded by the original model.
Impaired ability to alter synaptic strength in a physiological meaningful way can contribute to both hypokinetic (e.g., PD) and hyperkinetic (e.g., HD and LIDs) disorders, depending on the mechanism and location of the pathology. In a seminal study, Kreitzer and Malenka (Kreitzer and Malenka 2007) demonstrated that corticostriatal LTD is lost in iSPNs in a mouse model of PD, and pharmacologically rescuing LTD at these synapses (by increasing striatal D2 receptor activation and reducing endocannabinoid degradation) also ameliorates behavioral deficits. Recent work in mice containing mutant variants of the huntingtin gene demonstrated that corticostriatal LTP is selectively lost in iSPNs early on in disease progression (due to impaired BDNF signaling) (Plotkin and others 2014). While impaired LTP at corticostriatal synapses on iSPNs may contribute to a hyperkinetic symptomatology in HD, pathologically excessive LTP at dSPNs may underlie hyperkinetic behaviors in LIDs (Picconi and others 2003; Shen and others 2015). This is likely due to periods of prolonged activation of D1Rs following L-DOPA administrations (Cenci and Lindgren 2007). Both excessive LTP in dSPNs (in mice) and dyskinetic behaviors (in mice and nonhuman primates) can be corrected by enhancing activation of M4 receptors (which counterbalances the D1R mediated increase in PKA activity and promotes LTD; see Box 3), offering an elegant way to alleviate dyskinetic behaviors without sacrificing the benefits of L-DOPA treatment (Shen and others 2015).
Not all forms of plasticity involve LTP and LTD. Neurons employ homeostatic mechanisms to adapt to normal and pathological alterations in network activity and their local environment (Brickley and others 2001; Turrigiano and others 1998; Yu and Goda 2009; Zhai and others 2018). Mounting evidence suggests that many striatal circuit pathologies underlying PD stem from maladaptive homeostatic adaptations. Acute activation of D1Rs modulates a constellation of postsynaptic conductances that ultimately increases the excitability of dSPNs, while acute activation of D2Rs leads to a reduction in iSPN excitability (Gerfen and Surmeier 2011). After chronic dopamine depletion, however, SPNs adapt to the lack of excitatory or inhibitory modulation by increasing intrinsic excitability in dSPNs and decreasing it in iSPNs (Fieblinger and others 2014). Prolonged L-DOPA administration induces a further set of complex functional and morphological homeostatic adaptations that include pruning and addition of specific classes of synaptic inputs to SPNs (Fieblinger and Cenci 2015; Fieblinger and others 2014; Fieblinger and others 2018; Suarez and others 2016; Zhai and others 2018). Though the etiology of such changes may be driven by an attempt to retain a degree of normalcy in terms of activity, the changes in network connectivity driven by synapse pruning and formation may themselves contribute to pathology and the circuit-underpinnings of LIDs (Fieblinger and others 2014). Such homeostatic adaptations to diminished or elevated dopaminergic tone highlight the inherent limitation of a static circuit model.
Summary
The classic box-and-arrow model was a transformative advance that still holds enormous utility, but more recent advances in the field have shed light on its limitations. Decades of research have since uncovered complexities in intrastriatal processing that shape the engagement and patterning of the direct and indirect pathways, including antagonistic interactions between the pathways themselves. It has also become apparent that the striatum (and BG in general) is not a static network but is rather flexible and adaptive—pathophysiology is often the result of aberrant synchronous/rhythmic activity, synaptic plasticity or maladaptive compensations rather than generalized increases and decreases in the “strengths” of striatal output arrows (Fig. 4). Representing these attributes in a two-dimensional diagram is not a straightforward task but recognizing them is crucial in the search for the pathological loci that should be targeted to treat complex movement disorders while avoiding deleterious side effects.
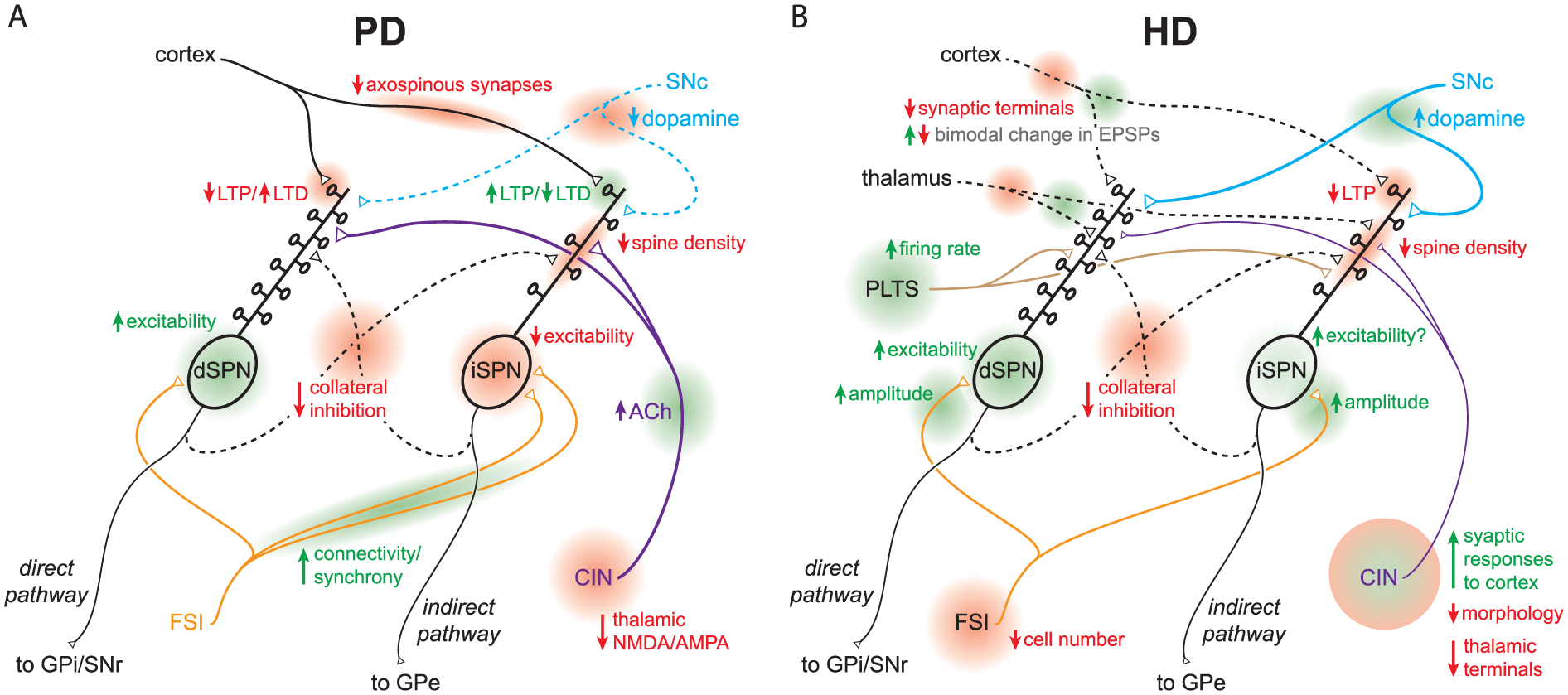
Current models of striatal circuit pathology in PD and HD. Diagrams focus on functional and anatomical microcircuit alterations; simplified and non-exhaustive due to space constraints. A. Circuit pathologies in PD. B. Circuit pathologies in early symptomatic HD, pre-SPN death. The diagrams highlight unique and complex constellations of striatal circuit alterations (not just elevated direct and indirect pathways) that sum to yield disease symptomatology. Loci of circuit alterations are highlighted in red/green and labeled. Abbreviations: see text.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by an NIH-NINDS grant (R01NS104089) to J.L.P., a European Research Council Consolidator grant (646880-SynChI) to J.A.G., and a US-Israel Binational Science Foundation grant (no. 2015255) to J.L.P. and J.A.G.