
Abstract
Inflammation plays a pivotal role in every stage of cancer development, from tumor initiation to progression, largely by influencing the balance between immune surveillance and immune evasion. At the heart of this inflammatory signaling is the transcription factor Nuclear Factor κB (NFκB), widely recognized as a key orchestrator of immune responses in malignancies. Within the framework of cancer immunoediting, comprising the three key phases Elimination, Equilibrium, and Escape, NFκB emerges as a double-edged sword. While it can enhance the activity and infiltration of effector immune cells that promote tumor destruction, it can also reprogram these cells into immunosuppressive subsets, thereby facilitating tumor immune evasion and growth. This dual functionality makes NFκB both a promising and challenging target in cancer immunotherapy. Despite its significance, comprehensive insights into NFκB’s roles across the immunoediting continuum remain limited. This review aims to bridge that gap by systematically exploring the multifaceted contributions of NFκB in modulating tumor-immune interactions, from immune-mediated tumor surveillance to immune escape, highlighting its potential and complexity as a therapeutic target.
Plain Language Summary
Inflammation has a key role in cancer’s development and spread. It influences whether the immune system can fight cancer or if the cancer escapes detection. NFkB, a protein that regulates the immune system, plays an important role in this process. Within the framework of cancer immunoediting, involving three important phases: Elimination, Equilibrium, and Escape, NFkB emerges as a double-edged sword. It can assist the immune system fight cancer, but it can also suppress immune cell function, allowing cancer to spread. NFkB is a complex target for cancer treatment due to its potential for both beneficial and detrimental effects. This review examines how NFkB influences the immune system's interaction with cancer at various stages, and how a better understanding it could lead to more effective treatments.
Introduction
Cancer is a complex, multi-step disease driven by the accumulation of genetic mutations, including the activation of proto-oncogenes and the inactivation of tumor suppressor genes. 1 The loss of tumor suppressor function plays a critical role by allowing cells to bypass natural aging mechanisms like senescence, enabling them to acquire an immortal phenotype that, when combined with further genetic alterations, can lead to malignancy. 2 However, genetic transformation alone is not sufficient. Emerging tumor cells must also evade the body’s immune system, which is equipped to recognize and eliminate abnormal cells. This vigilant immune activity initiates what is known as immune surveillance, a process that targets and destroys nascent tumor cells before they can establish disease.2,3 Despite this defense, a subset of genetically unstable tumor cells may survive immune elimination, especially their proliferation is temporarily kept in check by immune pressure. 4 Over time, these surviving cells may accumulate additional mutations that allow them to escape immune detection and thrive. 4 The capacity of tumors to evade immune responses is now considered a fundamental hallmark of cancer.
The concepts of cancer immunosurveillance and immunoediting demonstrated about the immune system’s role, not only in detecting and eliminating cancer cells but also its limitations. 5 Cancer immunoediting, a dynamic process that describes the dual role of the immune system in both suppressing and promoting tumor development. It encompasses three interconnected phases: elimination, where immune cells detect and destroy cancer cells; equilibrium, a state of balance in which residual tumor cells persist but are kept under control; and escape, where tumors acquire the ability to grow unchecked by the immune system. As tumors expand and progress to detectable stages, they begin to release soluble factors that further manipulate the tumor microenvironment (TME) to their advantage, contributing to immune evasion and disease advancement. The immunoediting model has reshaped our understanding of how the immune system interacts with cancer, highlighting both its protective and permissive roles. This paradigm is central to developing next-generation immunotherapeutic strategies aimed at tipping the balance back in favor of anti-tumor immunity.4-6
Chronic inflammation is now recognized as one of the defining hallmarks of cancer, with profound regulatory roles in tumor initiation, progression, and the suppression of anti-tumor immunity. 7 Among the key molecular players in this context is the proinflammatory transcription factor Nuclear Factor κB (NFκB), which has long been implicated in the development and advancement of cancer. 8 NFκB’s role in cancer is complex and context-dependent. In immune cells, it can act as both a promoter and suppressor of tumor development, depending on the stage and nature of the immune response. 9 During the early phases of tumor detection, particularly the elimination stage, NFκB activation via various receptor signaling pathways helps stimulate anti-tumor immunity through cytokine release and the activation of cytotoxic functions in immune cells. 10 However, as the tumor adapts and evolves, the role of NFκB begins to shift. In the transition from immune equilibrium to immune escape, NFκB contributes to a pro-tumorigenic environment by reprogramming immune cell behavior, often suppressing their anti-tumor functions. It also drives the expression of several tumor-promoting factors, including growth factors, vascular endothelial growth factor (VEGF), and matrix metalloproteinase-9 (MMP9), all of which support tumor growth, angiogenesis and metastasis. 9
In this review, we explore the dual role of NFκB in regulating immune cell behavior during three phases of cancer immunoediting. We examine how NFκB signaling intersects with key molecular and cellular events that govern immune surveillance, the elimination of transformed cells, and the eventual immune escape of malignant cells. Finally, we discuss the broader implications of targeting NFκB as a potential strategy for enhancing cancer immunotherapy and improving clinical outcomes.
An Overview of NFκB Activation
The NFκB family of transcription factors plays a central role in regulating immune responses, inflammation, and cancer progression. This family comprises five key proteins—p65 (RelA), RelB, c-Rel, NFκB1 (p50/p105), and NFκB2 (p52/p100), which function by forming various homo- or heterodimeric complexes.
11
All members share a highly conserved N-terminal Rel Homology Domain (RHD) of approximately 300 amino acids, which is crucial for dimerization, DNA binding, nuclear localization, and interaction with inhibitory IκB proteins.
12
Based on their ability to activate transcription, NFκB proteins can be categorized into two groups. Only RelA (p65), RelB, and c-Rel contain a C-terminal transactivation domain (TAD), which enables them to directly initiate gene transcription
11
(Figure 1). Notably, RelB is functionally distinct, requiring not only its TAD but also a leucine zipper domain at the N-terminus for full transcriptional activity.
11
The p105 and p100 proteins serve as precursors and are processed into p50 and p52, respectively. Interestingly, not all NFκB dimers are transcriptionally active; p50 and p52 homodimers, for instance, can suppress κB-dependent gene expression by competing with the active dimers for DNA binding or by recruiting transcriptional repressors such as histone deacetylases to gene promoters.
13
Structural overview of NFκB family transcription factors. The NFκB family consists of five key members: RelA (p65), RelB, c-Rel, NFκB1 (p50/p105), and NFκB2 (p52/p100). A shared feature among all these proteins is the presence of a Rel homology domain (RHD) at the N-terminal region, which facilitates DNA binding, dimerization, and interaction with inhibitory IκB proteins. Among them, only RelA, RelB, and c-Rel possess a transactivation domain (TAD) at their C-terminal end, which is crucial for initiating gene transcription. Notably, RelB contains a leucine zipper (LZ) motif within its N-terminal region, which contributes to its specific dimerization properties and functional roles in gene regulation
In their resting state, NFκB dimers are sequestered in the cytoplasm through interactions with IκB proteins.
10
These interactions are mediated by ankyrin repeat motifs within the IκBs, which bind to the RHD of NFκB proteins and prevent their nuclear translocation. NFκB transcription factors are activated through two major signaling pathways: the canonical (classical) and noncanonical (alternative) pathway.14,15 These pathways are triggered by distinct stimuli and regulate different aspects of immune function. The canonical pathway is typically activated by pro-inflammatory cytokines, antigen receptor signaling in B and T cells, and pattern-recognition receptors (PRRs) that detect microbial components.
16
Activation of this pathway relies on the IκB kinase (IKK) complex, which is composed of two catalytic subunits, IKKα and IKKβ, and a regulatory subunit known as NEMO (NFκB essential modulator or IKKγ). Upon activation, the IKK complex phosphorylates IκB proteins, targeting them for proteasomal degradation. This process frees NFκB dimers—most commonly p50/RelA or p50/c-Rel, which then translocate rapidly and transiently into the nucleus to initiate gene transcription11,17 (Figure 2). Activation pathways of NFκB signaling in response to diverse stimuli. NFκB-signaling is triggered by a wide array of extracellular signals through receptors such as Toll-like receptors (TLRs), the IL1 receptor, tumor necrosis factor receptor (TNFR), and antigen receptors like the T cell receptor (TCR) and B cell receptor (BCR). These signals predominantly activate the canonical NFκB pathway. A key event in this pathway is the activation of the IκB kinase (IKK) complex, which phosphorylates the inhibitory protein IκBα, marking it for proteasomal degradation. This degradation liberates the NFκB dimers, typically p50/p65 that then translocate rapidly from the cytoplasm to the nucleus to initiate gene expression. Figure created with BioRender.com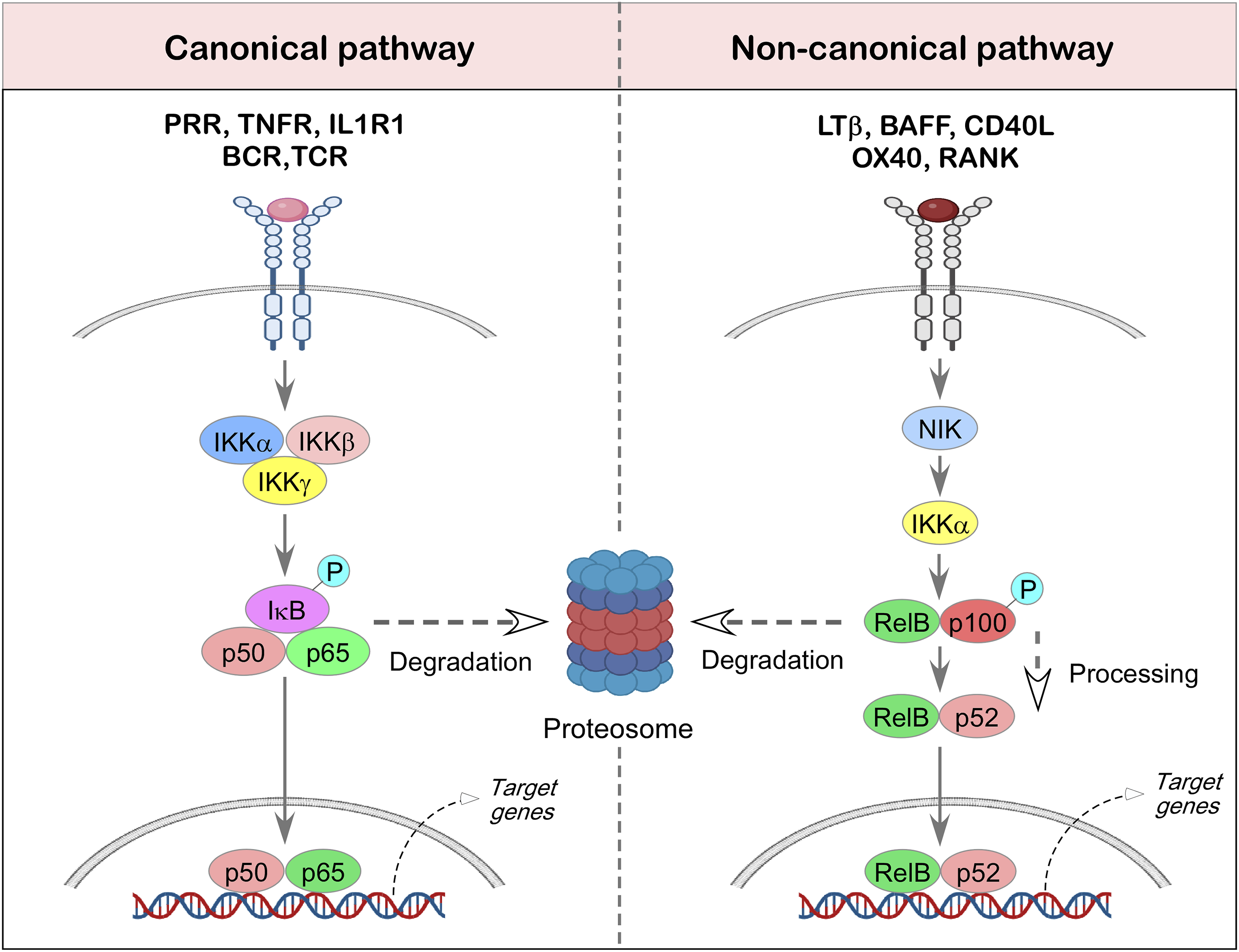
In contrast, the noncanonical pathway responds to a specific set of signals, primarily from members of the tumor necrosis factor receptor (TNFR) superfamily, such as lymphotoxin-β-receptor (LTβR), B-cell activating factor (BAFF) receptor (BAFFR), cluster of differentiation-40 (CD40), and receptor activator of nuclear factor κ-B (RANK). This pathway depends on the stabilization and activation of NFκB-inducing kinase (NIK), which in turn drives the processing of the p100 precursor into p52. The resulting p52/RelB dimers then translocate into the nucleus, where they modulate gene expression in a more sustained and regulated manner compared to the canonical route15,18 (Figure 2). Both signaling pathways play critical roles in shaping innate and adaptive immune responses within the tumor immune microenvironment. 10
NFκB in Tumor Microenvironment: From Immune Surveillance to Immune Evasion
Cancer is the battle between rapidly evolving tumor cells and the vigilant immune system. The Immune system recognize and eliminate cancer cells through a process known as immune surveillance, which relies on the detection of tumor-specific antigens.19,20 However, tumors often manage to persist and evolve despite this surveillance, a phenomenon explained by the concept of cancer immunoediting. The process describes how the immune system applies selective pressure, eliminating highly immunogenic tumor cells while inadvertently allowing less immunogenic variants to survive and proliferate
6
(Figure 3). Role of NFκB in the cancer immunoediting process. This schematic illustrates the dynamic interaction between the immune system and developing tumors, known as cancer immunoediting, which unfolds in three interconnected phases: elimination, equilibrium, and escape. In the elimination phase, both innate and adaptive immune cells actively recognize and destroy emerging tumor cells. NFκB plays a central role here by promoting the expression of pro-inflammatory cytokines and cytotoxic molecules such as perforin and granzyme, enhancing the immune system’s tumor-killing capacity. If complete tumor clearance fails, the process enters the equilibrium phase, where immune pressure limits tumor growth but does not eliminate all tumor cells. During this period, NFκB-signaling can influence immune surveillance while also contributing to the selection of tumor variants with reduced immunogenicity. Eventually, some of these variants may acquire the ability to avoid immune detection entirely, leading to the escape phase. Here, NFκB activity in tumor-associated immune cells may shift toward supporting tumor growth by suppressing effector immune responses and promoting a more immunosuppressive microenvironment. Figure created with BioRender.com
Adding to this complexity is the role of inflammation and its key regulator, NFκB. Chronic inflammation within the TME, often fueled by cytokines and chemokines produced by both tumor and immune cells, can significantly shape the trajectory of cancer. 21 NFκB, a central transcription factor in inflammatory signaling, has long been linked to tumorigenesis.8,9 It regulates genes involved in survival, proliferation, and metastasis, and its activation can tip the balance either toward tumor suppression or promotion. In immune cells, it can exert both pro- and anti-tumoral effects, depending on the context and cellular interactions within the TME.9,10,13 This dual behavior presents a significant challenge: why does NFκB drive tumor progression in some cases while supporting anti-tumor immunity in others?
To unravel this paradox, it is essential to address NFκB’s influence across a broad spectrum of immune cells. These include anti-tumor effectors such as T cells, B cells, dendritic cells (DCs), and natural killer (NK) cells, as well as immunosuppressive populations like tumor-associated macrophages (TAMs), myeloid-derived suppressor cells (MDSCs), T-regulatory (Treg) cells, and B-regulatory (Breg) cells.7,22 Each of these cell types plays a distinct role in shaping immune responses within the TME. Here, we’ve systematically explained the role of NFκB in different immune cells involved in phases of cancer immunoediting.
NFκB in the Elimination Phase
In this phase, the immune system mounts a coordinated defense to detect and destroy newly transformed tumor cells before they can establish malignancy. Cells such as NK cells, natural killer T (NKT) cells, and macrophages serve as first responders, identifying and attacking abnormal cells through direct cytotoxic mechanisms. In addition, they release pro-inflammatory cytokines that enhance immune vigilance and signal the need for further immune activation.4,5 A critical link between the innate and adaptive immune system is provided by dendritic cells. These antigen-presenting cells (APCs) internalize tumor antigens and migrate to nearby lymph nodes, where they initiate the priming of naïve T cells. This process leads to the activation of tumor-specific CD8+ cytotoxic T lymphocytes (CTLs) and CD4+ helper T cells, both of which are essential for mounting a targeted and effective anti-tumor response. 5 The NFκB signaling pathway promotes the expression of adhesion molecules that facilitate the recruitment of leukocytes to tumor sites, as well as cytokines that regulate immune responses, including TNFα, IL1, IL6, and IL8. 23 Constitutive activation of NFκB has been shown to enhance the production of chemokines by tumor cells, thereby supporting the development of anti-tumor immune responses. 23 Notably, tumor cell–intrinsic NFκB activity has been reported to play a critical role in lung tumor rejection, primarily by increasing the production of T cell–attracting chemokines and promoting the recruitment of CD8+ T cells.24,25 The NFκB signaling pathway plays a central role in regulating the development and function of various immune subtypes involved in this phase of cancer immunoediting.
NK and NKT Lymphocytes
NK and NKT cells are activated by inflammatory cytokines produced within the TME and play a frontline role in recognizing and destroying early tumor cells. 26 NK cells exert potent anti-tumor effects primarily through the release of cytotoxic molecules and cytokines like interferon-γ (IFNγ).27,28 This activity is largely dependent on the canonical NFκB signaling pathway, which is triggered by various activating surface receptors. Disruption of this pathway, such as through mutations in IκBα (an inhibitor of NFκB), has been shown to reduce NK cell proliferation. 29 Similarly, mutations in key NFκB pathway components like IKBKB or IKBKG impair IFNγ production, compromising NK cell functionality.30,31 NKT cells, which share characteristics of both NK cells and conventional T cells, also contribute significantly to early anti-tumor immunity. These cells can directly kill tumor cells and also modulate other immune cells by secreting a range of cytokines, including IFNγ, tumor necrosis factor-α (TNFα), interleukin-2 (IL2), IL4, IL5, and IL6. They promote tumor cell death via Fas ligand (FasL), tumor necrosis factor-related apoptosis-inducing ligand (TRAIL), perforin, and granzymes.22,32,47 NFκB signaling is essential for both the survival and differentiation of NKT cells. Study with animal models demonstrated canonical NFκB subunits have been linked to the expression of perforin (Prf1) and granzyme-B (Gzmb), key cytotoxic molecules required for tumor cell killing.33,34 Importantly, the release of tumor antigens following NK cell-mediated killing plays a critical role in priming T cells and strengthening the overall anti-tumor immune responses. 5
Innate Lymphoid Cells
Innate Lymphoid Cells (ILCs) are classified into ILC1, ILC2, and ILC3, with ILC1 being cytotoxic and ILC2/ILC3 functioning as helper cells. 35 IL15–activated ILC1s produce IFNγ, upregulating MHCI and inducing apoptosis, thereby activating tumor-specific immunity and suppressing early tumor growth.35-37 In hematological cancers, CD16-CD127+c-Kit-CRTH2-CD56+ ILC1-like populations secrete TRAIL and express cytotoxic markers (NKp30, NKp80), promoting tumor lysis.35,38 IL15 overexpression enhances TCR-NK1.1+CD49ahi ILC1-like cells, which release granzymes and anti-tumoral cytokines, limiting breast tumor progression. 35 Notably, NFκB directly regulates IFNγ, perforin, and granzyme expression, linking it to ILC-mediated cytotoxicity.39-41
ILC2s, activated by IL33 or IL25, recruit eosinophils and maintain immune balance in melanoma models, limiting metastasis.35,42 NFκB, particularly cRel, is essential for ILC2 effector functions, including CCL5 production, which recruits CD103+ dendritic cells and primes CD8+ T cells in pancreatic cancer.43,44 NCR+ILC3s contribute to anti-tumor immunity by producing TNFα, IL2, IL8, and IL22, important for early-stage lung cancer prevention.35,45 NFκB1 deficiency reduces ILC2 and ILC3 numbers, while NFκB1-RUNX1 interactions positively regulate ILC2 effector functions, underscoring NFκB’s central role in ILC-mediated anti-tumor responses. 46
γδ T Cells
γδ T cells exert anti-tumor effects through multiple mechanisms. Human Vγ9Vδ2 T lymphocytes kill tumor cells by releasing cytotoxic molecules such as granzyme and perforin and secrete pro-inflammatory cytokines via NFκB–dependent pathways.47-49 NFκB also contributes to the apoptotic resistance of γδ T cells. 50 CD16+ Vγ9Vδ2T cells mediate antibody-dependent cellular cytotoxicity (ADCC), functioning similarly to NK cells in tumor elimination. 47 Additionally, γδ T cells upregulate antigen-presenting molecules (HLA-DR), costimulatory and adhesion molecules (CD80, CD86, CD40), and scavenger receptor CD36 via MAPK and NFκB dependent pathways, further promoting tumor cell clearance.47,51-53 Despite these insights, the precise role of NFκB in γδ T cell biology warrants further investigation.
M1-Like Tumor-Associated Macrophages
M1-like macrophages, also referred to as classically activated macrophages, are key players in the immune system’s anti-tumor arsenal. They secrete pro-inflammatory cytokines such as IL1, TNFα, and IL12, which are crucial for activating various components of the adaptive immune system, including T cells, thereby enhancing the immune system’s ability to target and destroy cancer cells.54,55
The polarization of macrophages into the M1 phenotype is typically driven by myeloid differentiation primary response-88 (MyD88)-dependent toll-like receptor-4 (TLR4) signaling, with NFκB acting as a central transcription factor in this process. 56 One important downstream target is IL12B (IL12p40), which plays a vital role in the differentiation of T-helper-1 (Th1) cells, a subtype of T helper cells essential for effective anti-tumor immunity. 57 Moreover, M1 macrophages contribute to tumor cell killing by producing reactive oxygen species (ROS) and nitric oxide (NO), both of which have direct cytotoxic effects on cancer cells. 58 Additionally, these macrophages participate in ADCC, a mechanism that relies on the presence of tumor-specific antibodies to enhance immune-mediated tumor destruction. 59 NFκB signaling also regulates the expression of enzymes such as nicotinamide adenine dinucleotide phosphate (NADPH) oxidases, responsible for ROS generation,60,61 and inducible nitric oxide synthase (iNOS), which boosts NO production, especially in response to stimuli like LPS. Together, M1-like TAMs represent a potent force within the TME, contributing not only to direct tumor cell killing but also to the orchestration of broader immune responses against cancer.
Dendritic Cells
Dendritic cells act as professional APCs, not only capturing and processing tumor antigens but also producing cytokines that help orchestrate both innate and adaptive immunity. 62 Among the various DC subsets, type-1 conventional dendritic cells (cDC1s) are particularly important for anti-tumor responses. These cells specialize in cross-presenting tumor antigens to CD8+ T cells, leading to the secretion of IL12 and promoting strong cytotoxic T cell activity against cancer cells. 63 Similarly, cDC2s interact primarily with CD4+ T cells, further underscoring the role of DCs as a bridge between innate and adaptive immune defenses. 64
The maturation and function of these DC subsets are tightly regulated by NFκB-signaling pathways. Canonical NFκB components, activated through TLRs and CD40-signaling, are essential for DC maturation and activation. 65 The transcription factor c-Rel is critical for IL12 production in cDC1s, highlighting its key role in initiating T cell-mediated immunity.66,67 For cDC2s, stability and function depend on the non-canonical NFκB pathway, specifically the NIK/RelB– NFκB2 axis.68,69 RelB, a “non-canonical” NFκB protein, is required for the maturation of DCs into professional APCs. Interestingly, research by Shih et al. revealed that RelB activation in DCs can also occur through a RelB-p50 dimer rather than the typical RelB-p52 pathway, and this is regulated by canonical inhibitors such as IκBα and IκBε. 70 Once matured, dendritic cells migrate to tumor-draining lymph nodes, where they present tumor antigens to naïve T cells. This antigen presentation is pivotal for the activation of tumor-specific T cells, which then travel back to the tumor-site to eliminate cancer cells. 71
CD4+ and CD8+ T Lymphocytes
CD4+ and CD8+ T cells form the core of the adaptive immune response against cancer, working together to identify and eliminate tumor cells. NFκB-signaling has emerged as a key regulator of T cell activation, function, and survival within the TME.72,73 Among T cell subsets, cytotoxic CD8+ T cells and IFNγ–producing Th1 cells are especially vital for mounting effective anti-tumor immunity.74,75 Using CRISPR-Cas9 screening, Shifrut et al. demonstrated that RelA is essential for the proliferation of CD8+ T cells and their ability to produce IFNγ, a cytokine critical for tumor cell killing 76 (Figure 3). Similarly, NFκB-signaling plays a pivotal role in shaping the functional identity of CD4+ T cells by directing their polarization into specialized subsets. For example, RelA and c-Rel are involved in the differentiation of CD4+ T cells into Th17 cells, 77 which can contribute to tumor control by releasing IFNγ and by recruiting other immune cells, like DCs, NK cells, and CD8+ T cells, into the TME. 78 Another important subset, Th9 cells, has shown promise in anti-tumor responses. These IL9–secreting cells can promote tumor rejection by supporting T cell proliferation and inducing granzyme production. 79 Research indicates that NFκB components also regulate IL9 expression during Th9 differentiation. Specifically, TNFα–mediated activation of IKKα/β enhances this process, further linking NFκB to Th9-driven tumor immunity. 80
In CD8+ T cells, NFκB is indispensable for both their maturation and cytotoxic function. 81 Experimental models show that deleting Ikkβ or Rela specifically in T cells leads to weakened CD8+ T cell activity and accelerated tumor growth. 82 On the flip side, forced activation of IKKβ in T cells boosts their anti-tumor potential, demonstrating the therapeutic relevance of this pathway. 83 Once activated, tumor-specific CD8+ T cells migrate to the tumor-site, where they exert their cytotoxic effects through NFκB-regulated IFNγ production and direct cell killing 84 (Figure 3).
B Lymphocytes
B lymphocytes are commonly found within the TME, yet their exact contribution to cancer immunity remains a subject of ongoing investigation. B cells can exert notable anti-tumor effects through multiple mechanisms. Primarily, they produce tumor-specific antibodies that facilitate immune responses such as ADCC and opsonization, helping other immune cells recognize and destroy tumor cells. 85 Additionally, B cells can function as APCs, presenting tumor-derived antigens to CD4+ T cells and thus aiding in the initiation and amplification of adaptive immune responses.86,87
The NFκB signaling pathway is crucial for B cell biology, influencing their development, maturation, and antibody production. NFκB also regulates immunoglobulin class-switching, which is essential for generating effective, functionally diverse antibodies88,89 (Figure 3). Disruption of this pathway, particularly through the loss of key NFκB subunits like c-Rel or RelA, has been shown to impair B cell populations and diminish their functionality. Studies have reported that conditional deletion of these subunits in mature B cells leads to a reduction in their numbers, especially in the spleen.90,91 Moreover, the non-canonical NFκB-pathway, involving RelB and NFκB2, plays an important role in the formation and maintenance of germinal center (GC) B cells, which are central to high-affinity antibody production. 92 While the involvement of NFκB in T cell–mediated immunity is well established, the detailed mechanisms by which this pathway shapes B cell–driven anti-tumor responses remain less understood.
NFκB in the Equilibrium Phase
Following the initial immune attack in the elimination phase, some tumor cells manage to survive and enter the next critical stage of cancer immunoediting, the equilibrium phase. 4 This stage is characterized by a prolonged standoff between the immune system and the residual tumor cells, where active immune surveillance keeps the surviving tumor population in a state of functional dormancy. 4 During this time, the immune system continues to eliminate susceptible tumor cells, while new variants with reduced immunogenicity slowly emerge.5,93 These evolving tumor cell variants are shaped by ongoing immune selection pressure, primarily driven by the adaptive arm of the immune system.4,94 Unlike the elimination phase, where innate immunity plays a major role, the equilibrium phase is largely maintained by adaptive immune components, especially CD4+ and CD8+ T cells, as well as cytokines like IL12 and IFNγ. These molecules help sustain immune control over tumor cells, preventing their expansion while inadvertently promoting the outgrowth of immune-resistant clones.4,5
The delicate and dynamic balance between the immune system and tumor cells in the equilibrium phase is significantly influenced by cytokines, which either promote or restrain tumor progression. Among them, IL12 and IL23 play opposing roles in tumor immunity. IL12 supports anti-tumor responses, whereas IL23 appears to help tumors persist in a controlled, non-expanding state. In a fibrosarcoma mouse model, Teng and colleagues demonstrated that IL23 contributes to maintaining tumor cells in equilibrium. 95 Further studies revealed that the expression of IL23 within the TME is regulated by the p65NFκB subunit, emphasizing the role of NFκB in shaping immune responses during this phase. 96 IFNγ also applies immune pressure that keeps tumor growth in check. NFκB-signaling is instrumental in driving IFNγ production, linking this pathway to the prolonged immune surveillance observed during equilibrium. 97 Interestingly, immunosuppressive cytokines like IL10 have been implicated in sustaining tumor dormancy, further highlighting the complex interplay of immune signals during this stage. 98
Another critical factor is the balance between Th1 and Th2 cytokine responses. A shift in the Th1:Th2 ratio is thought to influence whether a tumor remains in equilibrium or transitions toward immune escape.99,100 NFκB subunits play a regulatory role in this context; p65 promotes Th1 cytokine IFNγ expression, while c-Rel supports the Th2 cytokine IL4. This cytokine balance may act as a molecular switch, determining whether the immune system continues to suppress the tumor or allows it to progress101,102 (Figure 3). Within the equilibrium tumor microenvironment, the presence of a robust population of effector immune cells, such as CD4+ and CD8+ T cells, is often coupled with a reduced number of regulatory immunosuppressive cells.98,103 However, when regulatory cells do infiltrate the tumor, they work to suppress inflammation and blunt the activity of effector cells, inadvertently shielding the tumor and allowing it to preserve its growth potential. 7 Thus, the ratio between immune-activating and immune-suppressing cells is a critical determinant of whether the tumor remains dormant or escapes immune control. 98 These findings underscore the importance of cytokine signaling and NFκB regulation in maintaining the equilibrium phase and suggest that even subtle shifts in this balance can significantly impact the trajectory of tumor development.
NFκB in Evading Immune Destruction
During the escape phase, tumor cells become less recognizable to the immune system and begin to exploit immune regulatory mechanisms to avoid elimination. What was once a hostile immune environment transforms into one that supports tumor survival and progression. In this phase, the TME becomes increasingly dominated by immune cells with tumor-promoting functions, including TAMs, MDSCs, Treg cells, and Breg cells. These cells contribute to tumor growth in two main ways: first, by suppressing the activity of anti-tumor immune cells such as CTLs, Th1 cells, NK cells, DCs, and M1-like macrophages104-106; and second, by secreting pro-tumorigenic factors like epidermal growth factor (EGF), VEGF to promote angiogenesis,107-109 and matrix-remodeling enzymes such as MMP9, cathepsins, and heparanase to facilitate invasion and metastasis110-112 (Figure 3).
This immune escape mechanism relies heavily on NFκB, which plays both tumor-intrinsic and extrinsic roles within the TME. NFκB directly regulates the transcription of several pro-tumorigenic molecules, including growth factors, VEGF, and MMP9, effectively linking chronic inflammation to cancer development and metastasis. 7 Extensive research has confirmed NFκB’s oncogenic potential. In murine models of K-Ras–driven lung cancer, for instance, conditional deletion of NFκB pathway components such as Ikkβ or RelA, or enforced expression of an IκBα super-repressor, significantly reduced tumor burden. Similar outcomes have been observed in other cancer types, including melanoma, reinforcing NFκB’s role in driving tumor progression.113-115 NFκB promotes breast cancer progression by regulating multiple oncogenic processes. It upregulates the anti-apoptotic protein Bcl2 and cyclin D1, supporting cell survival and proliferation.116,117 During carcinogenesis, PI3K–AKT–NFκB signaling drives epithelial–mesenchymal transition (EMT) by inducing N-cadherin and vimentin while suppressing E-cadherin, facilitating metastasis. 116 P65NFκB also regulates ZBTB7A, a biomarker that promotes EMT and metastatic potential, and stimulates VEGF expression, enhancing angiogenesis.116,118
NFκB contributes to chemoresistance by modulating ABC transporter proteins, including MRP1 and MDR1; its inhibition reduces their expression.116,119 AKT/NFκB–mediated GPR120 overexpression further drives ABC transporter activity, leading to epirubicin resistance. 120 Additionally, NFκB induces PDL1 expression on tumor cells, suppressing T cell activity and promoting tumor progression. 121 PDL1 can also be upregulated by inflammatory cytokines, such as TNFα, 122 and mediates intrinsic mechanisms that enhance tumor growth, metastasis, and resistance to FAS-ligand and chemotherapy-induced apoptosis, impairing anti-tumor immunity.121,123 Beyond these tumor-intrinsic effects, NFκB plays critical roles in various immune cell subtypes during the immune escape phase of cancer immunoediting, as discussed below. Understanding these regulatory networks offers promising avenues for therapeutic intervention aimed at reversing immune escape and re-engaging effective anti-tumor immunity.
M2-Like Tumor-Associated Macrophages
Tumor-associated macrophages are among the most abundant immune cells found within the TME, playing a pivotal role in shaping tumor fate. M2-like macrophages adopt an immunosuppressive, tissue-repairing profile that supports tumor growth and progression. 124 M2-like TAMs contribute to cancer development by secreting anti-inflammatory cytokines such as IL10 and transforming growth factor-β (TGFβ), along with chemokines, growth factors, and matrix-degrading enzymes that remodel the extracellular matrix and promote metastasis. 105 These macrophages help suppress effective immune responses by inhibiting the activity of DCs and T cells. For example, tumor-derived factors like soluble phosphatidylserine can skew macrophage polarization toward the M2 phenotype, dampening the anti-tumor immune response. 125
Emerging evidence links the NFκB-signaling pathway, particularly its canonical arm, to the pro-tumor activities of M2-like macrophages. Study revealed that NFκB, especially the p50 subunit, plays a central role in maintaining the M2 phenotype. 126 Inhibition of NFκB-signaling in TAMs, such as through targeted deletion of Iκb or RelA using the LysM-Cre mouse model, led to reduced tumor growth in models of cigarette smoke-induced lung cancer and inflammation-associated colon cancer. These findings highlight the potential of reprogramming M2-like macrophages back to the M1 phenotype by disrupting NFκB-signaling, thereby restoring their tumor-fighting capabilities.127,128 In several cancers, including breast, ovarian, and glioblastoma, canonical NFκB activation in macrophages has been shown to enhance IL10 production, reinforcing an immunosuppressive environment that favors tumor survival and immune evasion.129-131 Understanding this dual role of NFκB in M1 and M2-like macrophages offers new therapeutic opportunities to modulate macrophage function and disrupt the supportive niche that tumors create for themselves within the immune landscape.
Myeloid-Derived Suppressor Cells
Myeloid-derived suppressor cells have emerged as critical regulators of immune suppression within the TME. These cells are known for dampening anti-tumor immune responses by producing immunosuppressive factors such as IL10, TGFβ, and indoleamine 2,3-dioxygenase (IDO), which collectively help tumors evade immune destruction. 132 One of the key pathways involved in MDSC activation is the NFκB signaling cascade, particularly triggered by pro-inflammatory cytokines like IL1β. 133 This activation promotes MDSC expansion and enhances their suppressive function. Canonical NFκB-signaling appears to be central to MDSC biology. 134 Toll-like receptor-signaling, particularly through TLR2 and TLR4 engaging the MyD88 adaptor protein, has been shown to be necessary for optimal MDSC function in models of colon cancer and fibrosarcoma.135,136 Similarly, signaling through TNF receptors, especially TNFR2, further enhances MDSC-mediated immunosuppression, largely through NFκB-dependent IL10 production 137 (Figure 3).
Interestingly, while NFκB1-deficient mice (NFκB1−/−) exhibit an increased number of MDSCs in tumors, these cells are functionally impaired, resulting in a diminished ability to suppress immune responses and ultimately slower tumor growth. 138 Targeting specific NFκB subunits in macrophage-lineage cells has also revealed distinct roles: deletion of RelA using LysM-Cre-RelA-Flox mice reduces MDSC accumulation in glioblastoma models, 131 while c-Rel has been identified as a key regulator of MDSC development, function, and metabolism during tumor progression. 139 Together, these findings point to the multifaceted involvement of NFκB, through its subunits RelA, NFκB1, and c-Rel, in governing MDSC-mediated immune suppression. Understanding how NFκB orchestrates MDSC activity opens new avenues for disrupting this immune-evasive network and restoring effective anti-tumor immunity.
Innate Lymphoid Cells
Innate lymphoid cells 2 can exhibit pro-tumorigenic activity in cancers such as acute promyelocytic leukemia, prostate, and bladder malignancies by producing IL4 and IL13, which expand MDSCs and create an immunosuppressive microenvironment.140-142 ILC2-intrinsic cRel deficiency significantly reduces the expression of stimulatory receptors ICOSL and OX40L, as well as type-2 cytokines IL5, IL9, IL13, and GMCSF, highlighting the role of NFκB in regulating ILC2 effector functions. 43
In a mouse fibrosarcoma model, TGFβ-mediated NK-derived intermediate ILC1s (intILC1s) promoted tumor growth by producing platelet-derived growth factor (PDGF-AB), a pro-angiogenic factor, and expressing inhibitory receptors NKG2A and KLRG1. 143 Additionally, ILC2s with downregulated KLRG1 enhance CXCL2 and IL13 production, facilitating hepatocellular carcinoma (HCC) development through recruitment of immunosuppressive neutrophils.143,144 Notably, CXCL2 expression in HCC is regulated via NFκB signaling.145,146
γδ T Cells
IL17 produced by γδT17 cells promotes tumor proliferation by activating NFκB signaling and inducing tumor secretion of MMPs and VEGF, facilitating tumor invasion and metastasis. 147 γδT17 cell differentiation requires LTβR signaling and NFκB family members RelA and RelB: RelB downstream of LTβR drives RORγt and RORα4 expression, promoting thymic precursor differentiation into γδT17 cells, while RelA regulates LT ligand production in accessory thymocytes. 148 Tumor-infiltrating Vδ1 T cells secrete IL17 to recruit MDSCs and suppress CD8+ T cell activation. These Vδ1+ T cells also inhibit CD8+ αβ T cell cytotoxicity and dendritic cell function, further promoting an immunosuppressive TME.147,149
Tumor-Associated Immature Dendritic Cells
Within the TME, the presence of immature dendritic cells (iDCs) is often driven by tumor-secreted factors, such as VEGF. VEGF plays a critical role in recruiting immature myeloid cells from the bone marrow, which then accumulate as iDCs in the tumor milieu. 150 These iDCs not only fail to initiate effective immune responses but also actively contribute to immune suppression. They do so by producing immunosuppressive enzymes like IDO and arginase, which impair the function of circulating DCs and T cells.151,152 Additionally, VEGF interferes with the normal development and maturation of DCs by blocking NFκB-signaling in hematopoietic stem cells, weakening the body’s ability to mount a robust anti-tumor immune response. 153 Furthermore, it activates CD4+CD25+ Treg cells, which suppress T cell activation.154,155
T-Regulatory Cells
T-regulatory (Treg) cells, a specialized subset of CD4+ T cells identified by the markers CD4+CD25highCD127-FOXP3+, are essential for maintaining immune tolerance by preventing autoimmunity. However, their immunosuppressive nature also poses a challenge in cancer, where they can inhibit effective anti-tumor immune responses.106,156 Research has highlighted the pivotal role of NFκB-signaling pathway, particularly its RelA and cRel subunits, in regulating forkhead box-P3 (FOXP3) expression and supporting Treg development in the thymus via the PKCθ–CBM–IKK signaling axis.157-159 Experimental deletion of key NFκB signaling components—such as Carma1, Bcl10, Malt1, Ikb, or RelA, in Tregs using the Foxp3-Cre model leads to a marked reduction in Treg generation. This loss impairs the expression of key Treg-associated genes, elevates pro-inflammatory cytokines like IFNγ and TNFα, and compromises Treg functionality, which ultimately results in reduced tumor growth in transplantation models160-164 (Figure 3).
Treg cells suppress immune responses through both direct cell contact, via molecules such as cytotoxic T-lymphocyte-associated protein-4 (CTLA4), Lymphocyte activation gene-3 (LAG3), and T-cell immunoreceptor with Ig and ITIM domains (TIGIT), as well as by secretory mediators including IL10, TGFβ, IL35, and Gzmb 165 (Figure 3). Notably, c-Rel is crucial for the expression of TIGIT, and its deficiency results in downregulation of this inhibitory receptor. 166 Similarly, deletion of Nfkb2 in Tregs impairs their suppressive ability and causes systemic inflammation, with associated reductions in CTLA4 expression. Both pharmacological and genetic inhibition of c-Rel significantly impact the expression of essential Treg-associated genes like Gzmb and Tgfb1 in cancer settings.167,168 The recruitment of Treg cells into the TME is also enhanced by CCR4, a chemokine receptor whose expression is transcriptionally regulated by NFκB-signaling.169,170 While CD4+ Treg cells dominate, a smaller population of CD8+ Tregs has also been observed within tumors, where they contribute to immune evasion and tumor progression.106,171
B-Regulatory Cells
B-regulatory (Breg) cells are a specialized subset of B cells known for their immunosuppressive functions. These cells play a key role in maintaining immune tolerance, largely through the secretion of anti-inflammatory cytokines such as IL10 and TGFβ. 106 In the context of cancer, studies have shown that Breg cells are often expanded within the TME, where they contribute to immune evasion by suppressing the activity of CD4+ and CD8+ effector T cells and promoting the proliferation of Treg cells. 172 The development and function of Breg cells are closely linked to the TLR–MyD88–NFκB signaling axis. Activation of this pathway is essential for Breg differentiation, equipping them with immunoregulatory properties that dampen T cell responses and support tumor-induced immune suppression 173 (Figure 3). A critical player in this process is IκBNS, a nuclear IκB protein induced by TLR-signaling, which facilitates IL10 production in B cells. B cells lacking IκBNS show reduced expression of key transcription factors such as Blimp-1 and IRF4, along with diminished IL10 secretion, highlighting the importance of IκBNS in Breg-mediated regulation. 174 Further research has identified the phosphoinositide 3-kinase (PI3K)/AKT/NFκB signaling axis as instrumental in sustaining the immunosuppressive activity of PDL1+ Breg cells in tumor settings. Notably, inhibiting NFκB-signaling in Breg cells has been shown to restore anti-tumor immune responses in breast cancer models, pointing to its therapeutic potential. 175 Despite these findings, the complete picture of how Breg cells orchestrate immune suppression and facilitate tumor immune escape remains to be fully elucidated, underscoring the need for continued investigation in this area.
NFκB and Dysregulation of Hematopoiesis during Cancer
Hematopoietic stem and progenitor cells (HSPCs) give rise to all blood and immune cell lineages. 176 Increased circulating HSPCs, particularly granulocyte–monocyte progenitors (GMPs), multipotent progenitors (MPPs), and hematopoietic stem cells (HSCs), have been observed in patients across multiple cancers, correlating with immunosuppressive phenotypes, higher tumor grade, and poor prognosis.176,177 Within the TME, HSPCs undergo myeloid-biased differentiation, expanding tumor-associated myeloid populations such as MDSCs, neutrophils, and macrophages, driven by inflammatory cytokines including GMCSF, GCSF, IL6, and IL1. 178
Animal studies further support these observations. In MMTV-PyMT transgenic mice, expansion of HSCs, MPPs, and GMPs precedes the accumulation of immunosuppressive myeloid cells, mediated by GCSF.176,179 Similarly, mice bearing MC57 fibrosarcoma or B16-F10 melanoma exhibit HSPC expansion and myeloid-biased hematopoiesis.176,180 MDSCs suppress anti-tumor immunity through multiple mechanisms, with NFκB playing a key role in their tumor-promoting functions.131,181,182
In acute myeloid leukemia (AML), HSCs in leukemic bone marrow exhibit elevated early growth response-3 (EGR3), which induces cell cycle arrest and functional suppression; NFκB regulates EGR3 expression.183-185 Pharmacologic inhibition of NFκB selectively kills AML cells while sparing normal CD34+CD38- bone marrow cells, highlighting its importance in AML survival. 186 In chronic myeloid leukemia (CML), TME-derived TNFα activates NFκB/RelA signaling and enhances IL3 and GMCSF receptor expression, supporting leukemic stem/progenitor cell survival.186,187
Cancer-induced lymphopoiesis defects are also reported: B16-F10 melanoma–bearing mice show reduced circulating lymphocytes and decreased pre-B and immature B cells in bone marrow,176,188 while syngeneic EL4 thymoma models display declines in CLPs, NK precursors, NK cells, and B cells.176,189 In acute lymphoblastic leukemia (ALL), constitutive NFκB activation via RelA/p50 complexes promotes cell survival by enhancing proliferation and preventing apoptosis.186,190
Accumulating evidence indicates that dysregulated NFκB-signaling is deeply intertwined with both the development and maintenance of T-cell acute lymphoblastic leukemia (T-ALL) 191 . Early work by Kordes and colleagues demonstrated that NFκB is constitutively active in the majority of primary human T-ALL samples, with electrophoretic mobility shift assays detecting phosphorylated IκBα, active p50:p50 homodimers, and p50:RelA heterodimers in 11 out of 13 specimens. 190 These findings established that aberrant activation of the canonical NFκB pathway is a common biological feature of T-ALL.
Functional studies further support this notion. Pharmacological blockade of canonical IKK activity—through the NEMO-binding domain peptide, the proteasome inhibitor bortezomib, or the IKKβ-selective inhibitor BMS-345541, consistently suppresses NFκB-signaling and induces apoptosis in T-ALL cell lines.191-193 These observations underscore that T-ALL cells are not only exposed to persistent NFκB activity but are also dependent on it for survival. Mechanistic insights have also revealed cross-talk between oncogenic Notch signaling and the NFκB-axis. Notch promotes NFκB activation in T-ALL by driving ASB2-mediated degradation of IκBα, whereas targeted suppression of ASB2α decreases leukemic cell growth and triggers apoptotic death. 194
However, recent transcriptomic and developmental studies extend the relevance of NFκB far beyond its role as a survival factor. Early T-cell precursor ALL (ETP-ALL), a distinct, high-risk subtype derived from bone marrow–resident multipotent early thymic progenitors, shows markedly higher NFκB activation signatures compared with non-ETP T-ALL. ETP-ALL blasts also express elevated levels of genes associated with hematopoietic stemness. 195 These findings indicate that heightened NFκB activity is embedded within an early developmental context and may contribute to the persistence, plasticity, and transformation potential of immature progenitor-like leukemic cells.
This developmental perspective aligns with broader evidence showing that NFκB plays a fundamental role in maintaining stem-cell–like programs in several leukemia models. Beyond its classical function in promoting proliferation and survival, NFκB helps preserve primitive hematopoietic states that are inherently more susceptible to malignant evolution. 196 Thus, dysregulated NFκB not only supports established leukemic clones but may also shape the trajectory of leukemogenesis by perturbing lineage commitment, expanding progenitor pools, and sustaining stem-like transcriptional programs during early T-cell development.
The connection between NFκB-driven inflammation and hematologic dysregulation also extends to clonal hematopoiesis (CH). CH occurs at significantly higher frequencies in patients with solid tumors than in healthy individuals, and a high CH burden is associated with adverse clinical outcomes across cancers. 197 In non–small cell lung cancer (NSCLC), for example, patients with elevated CH loads show enhanced inflammatory activation within myeloid cells, a process mediated largely through NFκB signaling. 197 These observations reinforce that NFκB dysregulation is a shared axis linking inflammatory stress, expansion of aberrant hematopoietic clones, and malignant progression.
Collectively, these findings provide a more integrated understanding of NFκB biology in T-ALL and CH. Rather than functioning solely as a survival regulator in established leukemia, NFκB emerges as a developmental determinant that influences hematopoietic progenitor behavior, lineage specification, leukemic transformation, and the inflammatory milieu that sustains malignant evolution. This revised interpretation addresses the reviewer’s insight that NFκB contributes to both the ontogeny and persistence of leukemia, positioning it as a central node in the pathobiology of T-ALL and related hematopoietic disorders.
NFκB and Virus-Induced Cancer
Oncogenic viruses exploit NFκB-signaling to promote tumorigenesis.
198
In Epstein-Barr virus (EBV)–associated cancers, the latent membrane protein-1 (LMP1) activates NFκB, increasing expression of cell surface antigens (CD95, CD54, CD40), proinflammatory cytokines, angiogenic factors (COX2, VEGF), and anti-apoptotic proteins (BCL2, A20, cIAP, BFL1).198,199 In nasopharyngeal carcinoma, LMP1–mediated NFκB activation upregulates GLUT1 to enhance glucose uptake, supporting proliferation,
200
and promotes immune escape in NK/T cell lymphoma via PDL1 upregulation (Figure 4).
201
LMP1 also silences the tumor suppressor PTEN through NFκB –dependent DNA methyltransferase activation and enhances HIF1α activity, further driving tumor progression.202,203 NFκB in virus-induced oncogenesis. (A) Schematic representation of virus-mediated activation of the NFκB signaling pathway and its contribution to oncogenic transformation. (B) Summary of the major oncogenic mechanisms driven by viral activation of NFκB. Viral gene products interact with components of the NFκB cascade, resulting in the transcription of genes involved in chronic inflammation, tumor cell proliferation and survival, angiogenesis, metabolic reprogramming, immune evasion, and other tumor-promoting processes. The cumulative effects of these alterations facilitate malignant transformation, tumor progression, and metastatic dissemination. Abbreviations: EBV, Epstein–Barr virus; HPV, human papillomavirus; KSHV, Kaposi sarcoma-associated herpesvirus; HBV, hepatitis B virus; HTLV-I, human T-cell leukemia virus type-I. Figure created with BioRender.com
Kaposi sarcoma-associated herpesvirus (KSHV) activates NFκB via viral FLICE inhibitory protein (vFLIP), essential for viral latency, survival, and carcinogenesis in primary effusion lymphoma (PEL). 198 NFκB activation by vFLIP enhances IRF4-mediated interferon-stimulated gene expression. 204 KSHV viral G-protein coupled receptor (vGPCR) stimulates NFκB via TAK1, inducing IL8, COX2, BCL2, and VEGF, promoting tumor growth and angiogenesis (Figure 4).198,205
Human papillomavirus (HPV) oncogenes E6 and E7 manipulate NFκB to facilitate chronic inflammation and cervical cancer progression.206,207 These proteins modulate COX2–mediated prostaglandin synthesis, enhancing proliferation, angiogenesis, and inhibiting apoptosis. 206 E7 inhibits cytoplasmic kappa-B kinase, while E6 reduces NFκB p65–dependent transcription in the nucleus, suppressing IL1α, IL1β, and CXCL8/IL8 expression.207-210 HPV also promotes an immunosuppressive microenvironment by upregulating CXCL12/SDF1 to recruit FOXP3+ Treg cells and inducing IL10 and TGFβ expression.207,211,212
Hepatitis B virus (HBV) suppresses the innate immune responses for their survival, replication, as well as its carcinogenic manifestation. 213 Study demonstrated HBV damages the innate immune system by ubiquitination, in which the virus accelerates the ubiquitin-dependent degradation of numerous pattern recognition receptors and their downstream adaptors. MAVS, an adapter that enables the MDA-5/RIG-I-mediated immunological response, is the primary targets for ubiquitination by HBx. 214 According to the study, the 136th lysine residue on MAVS serves as a substrate for HBx’s degradative ubiquitination and dampening of the MAVS-dependent interferon response.214,215 Furthermore, HBx-induced K48-linked polyubiquitination degrades cGAS in a proteasome-independent way, leads to reduced IFN-I production.214,216
Conversely, HBx also removes K63-linked polyubiquitin chains from IRF3, IRF7, and TRAF3 (which are part of pattern recognition signaling pathways) from activating downstream effectors and spreading the pattern-mediated signaling. 214 Accordingly, HBx may promote or prevent the ubiquitination of particular proteins, which could compromise the type I interferon response. Similar to HBx, HBV polymerase damages the cytosolic dsDNA sensor STING by removing K63-linked polyubiquitin chains, which hinders STING’s capacity to trigger a type I interferon response. 217 Additionally, during HBV infection, the HBV e antigen accomplishes a similar function by binding to NEMO and protecting it from TRAF6-mediated K63-linked polyubiquitination, which significantly lowers NFκB activity and the production of proinflammatory cytokines. 218
The Human T cell leukemia virus type I (HTLV-I) Tax protein is thought to initiate and influence the pathogenesis of adult T cell leukemia (ATL). 219 Tax is an effective inducer of IκB degradation and NFκB nuclear accumulation. NFκB is required for the survival and immortalization of HTLV-I transformed cells. 219 According to the study, Tax requires NFκB to immortalize primary T cells since Tax mutants that do not activate NFκB cannot do so.219,220 Furthermore, NFκB activation is also required for the survival of HTLV-I-infected cells. Pharmacological inhibition of NFκB by BAY11-7082 treatment induces programmed cell death of HTLV-I transformed cell lines and ATL leukemic cells through downregulation of antiapoptotic gene BCLxL (Figure 4).219,221
Targeting NFκB as Therapeutic Strategy
Emerging evidence has highlighted the dual nature of NFκB in cancer. While this transcription factor can support immune surveillance in early stages, it often shifts roles during tumor progression, fueling immune evasion, chronic inflammation, and ultimately poor clinical outcomes. These insights have prompted growing interest in targeting the NFκB pathway as a therapeutic strategy in cancer treatment. Innovative approaches to target the NFκB pathway have demonstrated significant potential in enhancing anti-tumor immunity. One such strategy involves a dual pH-sensitive nanocarrier system co-delivering curcumin and anti-programmed cell death protein-1 (PD1) monoclonal antibodies, which not only blocks PD1+ T cells but also suppresses the activity of TAMs. This combined action leads to a more robust anti-tumor immune response.
222
NFκB plays a pivotal role in maintaining PDL1 expression and stability in tumor cells. In breast cancer models, curcumin, an NFκB inhibitor, has been shown to reduce PDL1 levels on tumor cells and promote infiltration of CD8+ cytotoxic T cells, thereby improving anti-tumor responses.223,224 Several other NFκB inhibitors, including mepazine, KINK1, and pentoxifylline (PTXF), have been effective in suppressing the immunosuppressive functions of MDSCs and Treg cells.139,162,164,166 These agents not only reduce the number of Treg cells but also destabilize their immunosuppressive phenotype, leading to the release of pro-inflammatory cytokines like IFNγ and TNFα within the TME,162,166,225,226 (Figure 5). In melanoma models, the selective inhibition of c-Rel, using R96A, significantly reduced the suppressive capacity of MDSCs
139
(Figure 5). Most importantly, many of these inhibitors lead to a boost in either the number or activity of CD8+ T cells, the primary effectors of anti-tumor immunity. Their increased presence and function within the TME are crucial for effective tumor elimination. Therapeutic targeting of NFκB in tumor immunity. Mechanistic illustration highlights the potential of targeting NFκB-signaling as part of cancer immunotherapy. In untreated conditions, Treg cells and MDSCs actively suppress the function of effector CD4+ and CD8+ T cells, enabling tumors to escape immune surveillance. However, therapeutic strategies that include NFκB inhibitors can disrupt this immunosuppressive environment. When combined with immunotherapy, NFκB inhibition reduces the activity of Treg cells and MDSCs, thereby enhancing the response of T-effector cells. This leads to higher production of IFNγ, a key cytokine involved in anti-tumor immunity, ultimately contributing to tumor regression. Figure created with BioRender.com
Experimental evidence underscores the potential of NFκB inhibitors in enhancing anti-tumor immunity, although their success is highly context-dependent. For instance, in RAG1−/− mice, which lack functional B and T cells, treatments with mepazine or PTXF fail to reduce tumor burden, indicating that the presence of adaptive immune cells is essential for these therapies to work effectively.162,166 This highlights that while NFκB inhibitors can improve tumor immunity; they are not universally effective across all patient populations or tumor types. The variability in therapeutic outcomes is believed to stem from two key challenges: an incomplete understanding of the molecular mechanisms behind patient response and resistance, and the risk of severe side effects associated with systemic NFκB inhibition. Importantly, the effectiveness of these inhibitors is closely tied to the nature of the tumor and its immune-microenvironment. Studies suggest that inflammation-driven cancers and NFκB–dependent tumors (“NFκB-addicted”) are more likely to respond favorably to this approach. 227
NFκB is a complex transcriptional regulator composed of multiple subunits, each exerting distinct roles in various cell types, including immune cells, tumor cells, and healthy tissue. 228 This intricacy means that indiscriminate targeting of NFκB can disrupt vital immune functions. For example, while NFκB supports immunosuppressive cells like MDSCs and Treg cells within the TME,162,164 it is also essential for the function of effector T cells that fight tumors.9,229 Therefore, careful consideration of dosage, timing, and cellular specificity is critical to minimize unintended consequences. To address this, researchers are developing antibody-drug conjugates to deliver NFκB inhibitors selectively to tumor cells or immunosuppressive immune cells, thereby sparing beneficial immune functions. 230 Moreover, combining NFκB inhibition with existing immunotherapies has shown promising synergistic effects. For example, curcumin combined with anti-CTLA4 therapy delays tumor progression in breast cancer models 223 (Figure 5). Similarly, agents like bortezomib, SMAC mimetics, mepazine, PTXF, or R96A have demonstrated enhanced efficacy when paired with anti-PD1 therapy in resistant tumors139,162,166,222,224,231,232 (Figure 5). Clinical studies have further validated this approach. In multiple myeloma, a combination of bortezomib and anti-PD1 has proven effective, offering new hope for patients with difficult-to-treat cancers. 231 Additionally, combining NFκB inhibitors with cytokine-based therapies, such as IL12 or IL18, has also enhanced anti-tumor responses in various tumor models.233,234
Understanding the dual role of NFκB in context of cancer has paved the way for the development of anti-NFκB therapies that not only suppress tumor-supportive immune cells but also reinvigorate cytotoxic immune responses. While challenges remain, particularly in optimizing safety and specificity, combining NFκB inhibitors with immunotherapies represents a promising strategy to reprogram the TME and improve clinical outcomes.
Conclusion and Future Perspectives
Over the years, substantial progress has been made in unraveling the complex relationship between the immune system and tumor development. Findings from both animal models and human clinical studies have revealed that the immune system not only defends the body against emerging tumors but can also contribute to tumor growth and immune evasion. This dynamic has led to the foundation of the concept of cancer immunoediting, which encompasses three phases: elimination, equilibrium, and escape.
This review has highlighted the pivotal and multifaceted role of NFκB in regulating immune responses during each of these stages. NFκB shows a clear dichotomy in its function: on one hand, it supports anti-tumor immunity through its action in effector immune cells such as NK cells, macrophages, CD4+, and CD8+ T cells, which are instrumental in recognizing and destroying early-stage tumors. On the other hand, its role becomes more complex as the tumor evolves. When the immune system fails to eliminate tumor cells, an equilibrium phase ensues, marked by a balance between immune control and tumor persistence. Over time, this can shift toward the immune escape phase, where the presence of immunosuppressive cells, such as Treg cells, MDSCs, and the pro-tumoral activity of NFκB contribute to diminished anti-tumor immunity and continued tumor progression.
A deeper understanding of the molecular and cellular mechanisms governing NFκB activation in various immune cell types could open up new avenues for therapeutic intervention. Targeting NFκB to promote tumor cell killing, while avoiding the unintended consequence of suppressing beneficial immune responses, remains a significant challenge. Despite the potential of NFκB inhibitors, their broad activity and associated toxicity have limited their clinical use.
To overcome these hurdles, precision-targeted strategies are essential. Future research should focus on dissecting the specific roles of individual NFκB subunits across the different stages of tumor progression. Conditional knockout/knock-in animal models, CRISPR-based genomic screening, and single-cell RNA sequencing will be crucial in mapping NFκB-regulated gene expression within distinct immune cell populations. Moreover, organoid systems that replicate the TME can serve as valuable platforms for high-throughput drug screening and functional studies. Ultimately, this knowledge will contribute to identifying selective modulators of the NFκB pathway, paving the way for safer and more effective immunotherapeutic strategies. By tailoring interventions based on the phase-specific and cell-specific roles of NFκB, we move closer to personalized and durable cancer treatments.
Footnotes
Acknowledgements
ORCID iDs
Author Contributions
SM did the background literature study and prepared the initial draft; SP prepared the figures; SD, TS, and SP contributed to the literature study and extended the initial draft; UB, SC literature study and arranged the references; GS supervised the entire project and made final editing to the draft.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: this study was funded by Indian Council for Medical Research, Government of India.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.