
Abstract
Autophagy is an ancient conserved and catabolic process required to maintain cellular homeostasis through sequestration and lysosomal degradation of cytosolic dysfunctional contents. The bulk degradation of cytosolic material through autophagy provides the necessary nutrients that cells utilize under starvation. Autophagy can play a mixed role in both the growth and survival of cancer cells. As a damaged cellular content-clearing mechanism, autophagy is involved in the early stage of tumor regression. Subsequently, in a mature tumor cell microenvironment, autophagy protects tumor cells from cellular stress, immune evasion, and therapeutic resistance. Therapeutic resistance mediated by autophagic regulators can make anticancer therapy less effective. This review discusses the molecular mechanism of mammalian autophagy, evaluates the dynamic role of autophagy in cancer, highlights autophagy-mediated therapeutic resistance, and suggests the use of biomarkers. In cancer therapy, the addition of biomarkers can provide specific identification, and the conjugation of biomarkers with chemo-drugs can reduce the side effects and ensure normal cell integrity.
Plain Language Summary
Plain Language Summary: This review explores the complex role of autophagy (a natural cellular recycling process) in cancer development and treatment resistance. While autophagy can suppress tumors in early stages by removing damaged components, it can also help mature tumors survive stress and evade therapy. The study details the molecular pathways of autophagy, highlights how it contributes to drug resistance, and proposes biomarker-based solutions to improve treatment precision. It underscores the need for new therapeutic strategies that target autophagy without harming normal cells, offering hope for more effective and less toxic cancer therapies.
Introduction
Cancer is a complex disease that has the second-highest global prevalence. 1 According to the World Health Organization (WHO) and the International Agency for Research on Cancer (IARC), an estimated 20 million new cancer diagnoses were reported in 2020, along with 9.7 million deaths worldwide. 2 The report also states that 1 in 6 individuals develop cancer in their lifetime. 3 This number is predicted to reach 35 million by 2050. 2 Due to uncontrolled cell growth and division, a mass or tumor is formed-characterized by rapid proliferation, high metabolic demands, and the induction of cellular stress. These consequences alter cellular conditions, often impacting fundamental survival mechanisms.4,5 Despite the heterogeneity in tumor etiology; like genetic and molecular underpinnings, phenotypic manifestation, and clinical presentations, 4 many human tumors exhibit dysregulation of autophagy-a crucial process in managing cellular stress and recycling nutrients. 6
Christian de Duve (a biochemist) marked the starting point of autophagy research through the discovery of lysosomes. 6 He first identified this organelle through the biochemical analysis of fractions obtained from the differential centrifugation of liver homogenates, characterized by its lytic function, and named it ‘lysosome’ in 1955. 7 Later in 1963, he proposed a concept of self-digestion process known as autophagy, 8 which degrades endogenous biomacromolecules to recycle with the aid of lysosomes. 6 This highly conserved mechanism evolved in ancient eukaryotes and is active at basal levels in all cells. 9 However, research on autophagy remained limited until the discovery of its core molecular machinery.10-12
Autophagy encompasses three main types (macro, micro, and chaperone-mediated autophagy); distinguished by the process of delivering cargo to the lysosome. 13 Macroautophagy (frequently referred to as autophagy) sequesters cytoplasmic material within an autophagosome prior to lysosomal fusion. 14 Microautophagy functions through the direct sequestration of cargo at the lysosomal membrane. 8 Chaperone-mediated autophagy (CMA) utilizes the chaperone Hsc70 to facilitate the degradation. 15 Autophagy’s function can be broadly categorized into 2 key aspects. Firstly, it acts as a cellular recycling system, breaking down biomacromolecules into glucose, free fatty acids, and amino acids for cell survival during starvation or stress. 9 Secondly, it regulates intracellular quality through the clearance of obsolete proteins, damaged organelles, unfolded DNA, toxins, and harmful microorganisms, facilitating the removal of dead cells along with longevity and self-recovery.16,17 Autophagy dynamically facilitates cell survival under metabolic stress.9,18 However, growing research has continued to uncover correlations between dysregulated autophagy and a range of human diseases, including aging, cardiovascular disease, neurodegeneration, inflammation, and cancer.9,17 For this review, we will mainly focus on the autophagic role in cancer cell regulation. Autophagy regulates the cellular mechanisms of cells that clear damaged proteins, organelles, and impaired DNA in the early stage of tumor formation, protecting normal cells. 19 The cytotoxic mechanism of autophagy can impede metastasis by preventing tumor cell necrosis and inflammation. 20 However, it has been demonstrated that in established tumors, autophagy enables tumor cells to cope with metabolic and nutritional stress, thereby facilitating their survival. 21
The dichotomous role of autophagy is crucial in cancer development. The critical link between impaired autophagy and cancer development was first established nearly 3 decades ago. Notably, in 1999, Levine’s group first correlated autophagy to cancer through identifying Beclin1 as a tumor suppressor. 22 Later, through a mouse model, they proved that heterozygous disruption of Beclin-1 can enhance tumorigenesis. 23 However, autophagy’s role is complex; within the tumor microenvironment, it can also protect tumor cells from various stressors and immune surveillance. 24 This duality contributes to significant therapeutic challenges. Despite decades of research, developing drugs that effectively target autophagy in cancer remains limited, largely due to autophagy’s contribution to therapeutic resistance. 19 Therefore, this review aims to elucidate the molecular mechanisms of autophagy in mammalian cells, examine its paradoxical roles in cancer pathogenesis and treatment response, followed by discussing the challenges of autophagy-mediated therapeutic resistance, and propose potential novel therapeutic strategies.
Methodology
This review synthesizes existing literature concerning the role of autophagy in cancer progression. The literature search encompassed articles published in English over the past 25 years, utilizing the PubMed, Google Scholar, and Science Direct databases. For selection, specific search terms and phrases were employed, including “autophagy dual role in cancer,” “autophagy in drug resistance,” “mammalian autophagy,” and “overview of autophagy.” The primary inclusion criteria prioritized systematic reviews, clinical trials, and other literature reviews that offered significant insights into the molecular mechanisms of autophagy in the context of cancer development (Figure 1). Overall, the search phrase “autophagy and cancer” on the databases mentioned above revealed an extensive body of literature published over the past 25 years, underscoring the importance of research in this field. Data Screening Illustration
General Aspects of Mammalian Autophagic Molecular Mechanisms
The discovery of ATG proteins marked a significant turning point in autophagy research. The substantial investigation into autophagic molecular mechanisms over the past 15 years has revealed the critical roles of autophagy in cell development, tissue homeostasis, metabolism, immune response, and various diseases. 25 The importance of these discoveries was underscored by the Nobel Prize awarded to Yoshinori Ohsumi in 2016 in Physiology or Medicine for his groundbreaking work in identifying core autophagic genes. 11 Eukaryotic cells utilize this highly catabolic process under diverse stressors such as hypoxia, heat stress, accumulation of ROS, energy or nutrient deprivation, ER stress, DNA damage, growth factors, and infection.17,26,27 Notably, amino acid deprivation has been the most extensively studied among these. 28 The molecular process of autophagy is delineated by sequential stages: initiation, phagophore assembly, autophagosome formation, subsequent autolysosomal degradation, a sequence frequently modulated by autophagy activators. 26
In mammalian cells, the phagophore, an isolation membrane, represents the initial morphologically identifiable structure formed de novo during autophagy, distinguishing it from pre-existing organelles.50,51 While it is established that the phagophore originates in close proximity to the ER at a subdomain termed an omegasome, the membrane curvature necessary for its formation is derived from various organelles, including mitochondria, the Golgi apparatus, and the plasma membrane.
52
Furthermore, a functional ATG cohort is recruited to the membranes to initiate autophagy. In addition, 40 ATG genes have been discovered to date.
53
Autophagy initiation requires exposure to diverse stressors such as nutrient deprivation, hypoxia, or growth factor deprivation, which trigger the dissociation of the mTORC1 from the ULK1 complex (a process mediated by dephosphorylation).30,54 However, AMPK functioning as a cellular energy sensor, upregulates autophagy through 2 main mechanisms: direct phosphorylation of ULK1 at Ser555 residues
55
and indirect activation of ULK1 by negatively regulating mTOR via TSC1/2
56
(Figure 2). Notably, AMPK and mTORC1 function as key regulators of autophagy, converging on the ULK1 complex to modulate its activity. This ULK1 complex, comprising ULK1/2, ATG101, ATG13, and the FIP200 kinase, is recognized as the primary initiator of the autophagy pathway.
30
The Mammalian Autophagic Process Inside the Cytosol. Mammalian Target of Rapamycin Complex1(mTORC1), AMP-Activated Protein Kinase (AMPK), Unc-51-like Kinase Complex (ULKc), Class III Phosphatidylinositol 3-Kinase (PI3KC3), Phosphatidylinositol 3-Phosphate (PI3P), Endoplasmic Reticulum (ER), WD-Repeat Protein Interacting With Phosphoinositide (WIPI), Double FYVE Domain-Containing protein1 (DFCP1), Vacuole Membrane Protein (VMP1), Transmembrane Protein 41B (TMEM41 B), Endosomal Sorting Complexes Required for Transport (ESCRT), Soluble N-ethylmaleimide-sensitive Factor Attachment Protein Receptor Complex (SNARE), Microtubule-Associated protein1A/1B-Light Chain-3 (LC3), Phosphatidylethanolamine (PE), Ubiquitin-like (UBL)
Functions of Mammalian Autophagic Regulators

The mature bilayer autophagosome formation from a phagophore requires 2 ubiquitin-like ATG conjugation cascades. 9 In the first conjugation cascade, a heterotrimeric complex of ATG12-ATG5-ATG16L forms by associating activated ATG12-ATG5 with ATG16L. 57 The initiation of this UBL cascade is mediated through 2 ubiquitin-activating enzymes (E1 and E2) of ATG7 and ATG10, respectively. 57 Additionally, WIPI2 associates with ATG12-ATG5-ATG16L and translocates it to the outer membrane of the phagophore, where a secondary cascade occurs. 63 The second pathway of the UBL conjugation cascade requires LC3. 67 The nascent LC3 undergoes cleavage by cysteine protease to form a conjugation with PE, mediated by E1, E2, and ATG12-ATG5-ATG16L super molecular complex hierarchically. 20 The lipidated LC3-PE (LC3-II) is tagged to the outer and inner membranes of the phagophore, providing selective cargo transportation, phagophore expansion, and closure. 57 In addition, LC3-II is also known as an autophagy biomarker, along with SQSTM1/p62 protein. During the initiation of autophagy, damaged proteins or dead cell aggregates within the cell are ubiquitinated. 30 This ubiquitination serves as a recognition signal for LC3-II and p62, which then facilitate the engulfment and subsequent delivery of these cytosolic materials to the autophagosome, respectively.30,68 After engulfing the cytosolic materials, the two ends of the membrane vesicles seal through the translocation of Vps37A to the phagophore, where it promotes CHAMP2-VPS4 assembly, a known ESCRT machinery. 69 This CHAMP2 and VPS4 assembly is required for bilayer autophagosome formation.36,66
In the final stage of the autophagy process, mature autophagosomes fuse with lysosomes to recycle cytosolic metabolites and produce amino acids, nucleic acids, fatty acids, and energy (Figure 2). Antecedent to this direct fusion, studies have shown that the outer membrane of the autophagosome can also fuse with early or late endosomes, forming an intermediate compartment known as the “Amphisome”. 57 Subsequently, the amphisome fuses with a lysosome to generate the autolysosome. Amphisomes formation is considered an obligate process to facilitate further lysosomal degradation presumably through the Rab family of small GTPases, tethering factors, Phosphoinositides, and SNARE proteins. 66 In the Rab family, specific GTPases recruit tethering factors that act as molecular bridges, stabilizing the lipid bilayers and facilitating the recruitment of SNARE proteins, which directly drive membrane fusion between the autophagosome and the lysosome. 70 Several other functional molecules, VAMP3 and VAMP7 have been implicated in membrane fusion. 71 However, recent studies have expanded our understanding by revealing the role of LC3-II in the assembly of this fusion machinery. 72 At the final stage of the autophagy process, recycled metabolites activate mTOR association with ULK1 along with ALR (Autophagic lysosome reformation) activation to terminate autophagy. 73
Cell Death Programs in Cancer: Apoptosis and Autophagy—Two Sides of the Same Coin
The death and survival programs in cancer cells are intertwined such that apoptosis and autophagy are positioned at opposite ends of a paradoxical bidirectional axis. Apoptosis is a caspase-dependent, energy-requiring program characterized by outer mitochondrial membrane permeabilization, cytochrome-c release, and executioner caspase activation, which culminates in immunologically quiet cell clearance. 74 This pathway is an important checkpoint against malignant progression in early oncogenesis and is a primary target of silencing by BCL-2 over-expression, mutant TP53, or death-receptor disorders in many cancers. In many respects, autophagy (and specifically macroautophagy) cuts across this system. Therefore, on one side of the coin, basal autophagy maintains proteostasis, removes defective organelles and genomic DNA damage, and limits oxidative stress, inhibiting the onset of cancer. 75 In the second scenario, the established tumor uses stress-induced autophagy to neutralize the metabolic burden, clear up ROS, and evade apoptosis in response to therapy, where autophagy mediates resistance to treatment and so-called metastatic fitness. 76
Mechanistically, apoptosis–autophagy crosstalk is bidirectional. Anti-apoptotic BCL-2/BCL-xL binds Beclin-1, inhibiting PI3KC3 complex formation and dampening autophagic flux; conversely, stress-activated caspases cleave Beclin-1, generating fragments that amplify mitochondrial apoptosis, converting a survival process into death signaling. 77 p53 coordinates both programs: nuclear p53 promotes autophagy indirectly (eg, TSC2/AMPK-ULK1 axis) and primes apoptosis, while cytosolic p53 can repress autophagy. Context is important here in this aspect. In nutrient-poor or hypoxic tumor regions, autophagy defers apoptosis (and necrosis) by recycling substrates and enabling ATP homeostasis; under overwhelming damage, autophagy can facilitate autophagy-dependent cell death (ADCD) by excessive self-digestion or through selective organelle removal that lowers the apoptotic threshold. 78 Therapeutically, this dichotomy argues for several stage-specific strategies: augment autophagy in premalignant/early lesions to enforce quality control and senescence; inhibit autophagy in advanced, therapy-refractory disease to disarm apoptosis evasion—ideally under biomarker guidance (LC3-II, Beclin-1, p62) and with longitudinal liquid-biopsy monitoring to capture temporal shifts.
Autophagy in Cancer: A Dichotomous Phenomenon
Autophagy is an important part of the quality control system and is implicated in cellular defense. It can also be described as a stress management system; whenever cells face particular stress or stimuli, they immediately upregulate autophagy to protect cells from deleterious effects. 79 The significant reduction of Beclin-1 expression in diverse human cancers, including human breast, ovarian, prostate, brain, lymphoma, and hepatocellular carcinoma, implies autophagy as a potential tumor antagonist.80-82 Conversely, in tumorigenesis, activation of autophagy provides survival and proliferation benefits to overcome the increased demand for tumor progression. 83 Hence, this tumor-enhancing activity of autophagy became one of the reasons for therapeutic ineffectiveness.
Autophagy Functions as a Tumor Suppressor
Autophagic Key Regulator
Beclin-1 is the first identified haploinsufficient tumor suppressor, monoallelically lost in several cancer cell lines.82,84 For example, tumor recurrence in human HCC tissues correlates with decreased beclin-1 expression. 85 Subsequent studies demonstrated the heterozygous disruption of beclin1 in a mutant mouse model that developed spontaneous HCC, 86 whereas mRNA downregulation of beclin-1 facilitates poor prognosis in human breast cancers. 87 Further research demonstrated the tumor suppressor activity of beclin-1 associated with UVRAG, which further enhances autophagy to prevent colon cancer. 88 Another study demonstrated that increased adrenaline activity can disrupt Beclin-1/hVps34/ATG14 complex functionality in autophagy to establish hepatocarcinogenesis.89,90 Considering its importance in tumor inhibition, beclin-1 has been studied most frequently.
PTEN is another tumor suppressor gene that acts as an inhibitor of PI3K/AKT/mTOR signaling pathway through inducing autophagy.91,92 PTEN functions as a PIP3 phosphatase, which can transform PIP3 to PIP2 by dephosphorylation to reduce PI3K activity
93
(Figure 3). Thus, negative regulation of PI3k restricts all the downstream signal transduction of the PI3K/AKT pathway, thereby promoting autophagy.
91
Recent studies demonstrated that in 30%–50% of prostate cancers, aberrant upregulation of the PI3K/AKT/mTOR pathway occurs due to deletion of PTEN.
94
In HT-29 colon cancer cells, significant autophagic activity is demonstrated upon PTEN activation.
95
PTEN regulation can be directly correlated with p53 interaction, as direct binding of p53 with PTEN promoter regions significantly upregulates PTEN mRNA expression to facilitate PTEN activity at the protein level.
91
Therefore, PTEN mutation or deletion remains among one of the defensive mechanisms of tumor cells. Tumor Repression Mediated by Autophagy Through Different Pathways. Autophagy (Ag), Reactive Oxygen Species (ROS), Phosphatase and TENsin Homolog Deleted on Chromosome 10 (PTEN), UV Radiation Resistance-Associated Gene (UVRAG), Genomic Stress (GS), Tuberous Sclerosis Complex 2 (TSC2), DNA Damage-Regulated Autophagy modulator1 (DRAM1), Sestrin1/2 (SESN1/2), Protein Phosphatase 2A (PP2A), Myelocytomatosis Oncogene (MYC), and Phosphate (P)
Autophagy and Senescence
Oncogene-induced senescence (OIS) describes an essentially irreversible arrest of proliferation in damaged or stressed cells in response to a mitotic burst following oncogene hyperactivation.96,97 OIS is a part of the natural barrier towards cellular transformation. 98 This process, generally coupled to a senescence-associated secretory phenotype (SASP), contains growth factors, proteases, bioactive lipids, chemokines, and cytokines; the majority display pro-inflammatory activity. 99 The cellular senescence program is considered a potent anti-proliferative mechanism, as the establishment and maintenance of this permanent cell arrest is governed by p53, p21, p16 INK4a, and pRB. 97 Autophagy can be an OIS effector, as delayed senescence is observed in ATG5 or ATG7 knockout cells, which inhibit IL-6 and IL-8 secretion. 84 Of note, IL-6 and IL-8 facilitate senescence activity in a cell-autonomous manner. 100
Another study demonstrated that, downregulation of ATG5/ATG7 prevents human diploid fibroblasts from undergoing senescence. 101 The finding suggests that cellular senescence mechanism can facilitate autophagy-dependent tumor dormancy. 102 Through intracellular recycling, autophagy provides amino acids for the synthesis of secretory proteins IL-6 and IL-8 to facilitate tumor growth inhibition. 84 Autophagy and senescence share some regulators, including p53, oxidative stress, and mTOR. 103 Among the regulators, p53 can regulate NK cells through the secretion of chemokines CCL2, and activated NK cells are surveilled through MHC-I for the removal of senescent cancer cells.97,104 In this context, blocking the autophagic gene following oncogene hyperactivation restricts permanent proliferative arrest of damaged cells, underscoring the anti-proliferative mechanism of autophagy.
Autophagy and Tumor Cell Necrosis
Cell necrosis, a passive form of unprogrammed cell death, occurs under different stimuli caused by acute cell dysfunction (change in oxygen level and temperature, radiation, electric shock), infections, ATP depletion, pathogens, toxins, and ischemia.105,106 Necrosis is considered an unregulated pathological process that can simultaneously affect a large number of cells without energy on a biochemical level. 106 Necrosis results in complete irreversible membrane damage, releasing inner cellular components into the extracellular space, which can provoke the immune system and facilitate inflammation. 106 Even though necrosis is considered a protective mechanism of cells from environmental stimuli, subsequently, different studies show that the excessive inflammatory compounds can further promote tumor growth. In this respect, 1 study demonstrated that in response to metabolic stress, cells undergo necrosis, while cells are incompetent for apoptosis and the autophagy process.78,107
Additionally, inflammatory cells, such as cytotoxic T-cells and NK cells, migrate to necrosis sites, facilitating pro-tumor immunity. 108 Another study demonstrates that macrophage production and pro-inflammatory cytokine production are associated with oncogene-activated necrosis to facilitate tumor growth. 78 Evidence suggests that blocking apoptosis via the Bcl-2 family may delay necrosis-associated tumor-inflammation by allowing autophagy. 78 Notably, hypoxia and cell necrosis are considered key differences between normal cells and tumor cells, 106 even though under hypoxic conditions, NF-κB activity is decreased, leading to autophagic regulator BNIP3 activation, inducing mitophagy to restrict cells from necrosis. 109 Inhibition of tumor cell necrosis is considered an effective tumor-prevention mechanism of autophagy.
Mechanism of Detoxification
The oncogenic proteins’ removal or dysfunction through autophagy is characterized by a detoxification mechanism. 36 Among oncogenes, c-MYC (Cellular Myelocytomatosis oncogene), a transcription factor, is dysregulated in ∼50% of human cancers. 110 The fundamental function of c-MYC in cell biology includes cell proliferation and differentiation, biomass accumulation, energy production, and cell size regulation. 110 Therefore, dysregulation of c-MYC contributes to tumor formation. It has been reported that a significant level of CIP2A protein was measured in a patient diagnosed with CML. 111 CIP2A, as a PP2A regulator, can inhibit PP2A-associated dephosphorylation of c-MYC, which facilitates c-MYC stability and increased oncogenic activity.111,112
In established cancers (gastric, ovarian, hepatocellular, NSCLC, and chronic leukemia), overexpression of CIP2A was observed clinically. 112 It has been reported that autophagic protein Ambra1 can modulate c-MYC levels in cells by dephosphorylation of c-MYC through promoting PP2A 113 (Figure 3). In addition, c-MYC has a pivotal role in immune cells; for instance, deletion of MYC in macrophages may restrict tumor growth through blocking pro-angiogenic molecules (VEGF, HIF-1α, and TGF) activity. 114 Notably, small increases in c-MYC levels can promote leukemia proliferation. 110
Limiting Oxidative Stress
Oxidative stress can be defined as a disparity between ROS and their eliminatory mechanism of antioxidant production. 115 Under normal physiological conditions, ROS levels will be balanced through antioxidants in due course. 105 However, infection through pathogens or stressors can produce excessive ROS that cannot be cleared by antioxidants and will eventually induce oxidative stress. 105 Stable ROS is necessary for cells as it can actively facilitate anti-inflammatory and antibacterial activities. 105 It has been reported that low levels of ROS can enhance cell growth and survival, conversely, excessive ROS levels are deleterious for cells as they can lead to ageing and diseases like cancer.105,116 The mitochondrial respiratory chain remains the principal source of reactive oxygen species, including hydrogen peroxide (H2O2), superoxide anion (O2-), hydroxyl radical (OH-), and organic peroxides.117,118 Oxidative stress induction can lead to cell death through autophagy, apoptosis, and necrosis. 105
Alternatively, ROS presence can promote autophagic machinery. 116 H2O2 (as an ROS compound) can facilitate carcinogenesis by effectively diffusing throughout mitochondria and the cell membrane, resulting in cellular injury. 115 It has been reported that induction of NF-κB can occur in the presence of H2O2, through a stepwise manner where some mediators like NIK (NF-κB-inducing kinase) and IKK activation, leading to the degradation of I-κB (NF-κB inhibitory kinase), resulting in regulation of NF-κB activity. 105 Activated NF-κB can induce the expression of anti-autophagic activity of Bcl-2 by interacting with Beclin-1. 105 NF-κB modulates cell proliferation and angiogenesis, additionally, downregulation of NF-κB blocks tumor cell proliferation.119,120 Other studies demonstrated inhibition of PTEN, activating Akt in the presence of ROS to facilitate tumor cell survival. 121 Overall, the activation of autophagy in the presence of oxidative stress is context-dependent, and ROS levels in cells can be a triggering point for future research in autophagy-dependent cancer treatment.
Genomic Instability
In response to genomic stress, p53 (the guardian of the genome) activates to maintain genomic stability. P53 can facilitate DNA replication errors to protect the genome, preventing any kind of cell abnormalities from malignant transformation through different mechanisms. 122 P53, as an important tumor suppressor, has been mutated in 42% of cases of different tumor cells, as proven through genomic sequencing. One regulated mechanism of p53 is to suppress tumors through upregulation of TGM2, which enhances autophagic activity in primary mammalian epithelial cells. 123 TGM2 is considered a biomarker for breast cancer, as upregulation of TGM2 correlates with poor prognosis in breast cancer. 124 P53 can induce autophagic machinery differently, for example, inhibiting the mTOR signaling pathway, a major endogenous inhibitor of autophagy 122 (Figure 3). The inhibition of mTOR can occur through direct upregulation of TSC2 by p53, and promote autophagy. 125 Another process described is the activation of AMPK, which can phosphorylate TSC1/2 and eventually inhibit mTOR activity and facilitate the initiation of autophagy. 49
Additionally, p53 target protein like Sestrin1/2, DRAM, and DAPK was shown to facilitate autophagy. DAPK, a potential tumor suppressor protein, remains silent in most malignant tumors through methylation at the promoter region. 49 Furthermore, DAPK1’s tumor suppressive activity depends on p53. Activated DAPK1 can phosphorylate Beclin-1 at threonine 119 residues to facilitate PI3KC3 complex formation to initiate phagophore formation. 126 Following genotoxic stress, AMPK phosphorylation and activation can be elicited by Sestrin1/2. 127 DRAM, an ancient lysosomal protein, can also facilitate p53-mediated autophagy induction. 49 As a lysosomal protein, DRAM can enhance autophagosome-lysosome fusion to recycle damaged cytosolic materials. Notably, p53 can also undergo senescence through activation of ATM in UV-induced DNA damage cells. Recovery from genomic stress is one of the most crucial parts of normal cell activity. 128 P53, as a caretaker of the gene, protects gene stability, but mutations often occur in this gene, which can lead to carcinogenesis.
Autophagy Functions as a Tumor Promoter
In mature tumors, metabolic stress and oxidative stress are constantly increased. To survive and fulfill these high metabolic and energy demands, autophagy is activated. This protective form of autophagy (pAg) will provide an alternative avenue for tumor survival. Here, we will describe some important regulatory factors that contribute to this pAg mechanism, including BNIP3, HMGB1, p62, AMBRA1, gene deletion, and chemoresistance (Figure 4). It has been reported that gene deletion of ATG5 and ATG7 can make cancer cells resistant to chemotherapeutic drugs. For example, in glioblastoma cells, HIF-1α induced autophagy to become resistant to the chemotherapeutic drug bevacizumab.
129
Additionally, ER-positive breast cancer cells obtain resistance to tamoxifen through the deletion of ATG5 and ATG7.
130
Resistance against cisplatin in ovarian cells is also facilitated by ATG5 deletion.
131
Moreover, mutation or deletion of key autophagic regulators, such as ATG5, ATG7, LC3, and Beclin-1, results in increased resistance of tumor cells against radiotherapy and chemotherapy.
20
Resistance against chemo or radiotherapeutic drugs makes the cancer treatment vulnerable. Tumor Promotion is Mediated by Autophagy Through Different Key Regulators of Autophagy. High Mobility Group Box1(HMGB1), Extracellular Signal-Regulated Kinase/Mitogen-Activated Protein Kinase (ERK/MAPK), Sequestosome1(p62/SQSTM1), TNF Receptor-Associated factor6 (TRAF6), Nuclear Factor Kappa B (NF-κB), Bcl-2 and Adenovirus E1B19-KDa-Interacting protein3 (BNIP3), Activating Molecule in Beclin-1-Regulated Autophagy protein1 (AMBRA1), and Normal Cell (NC)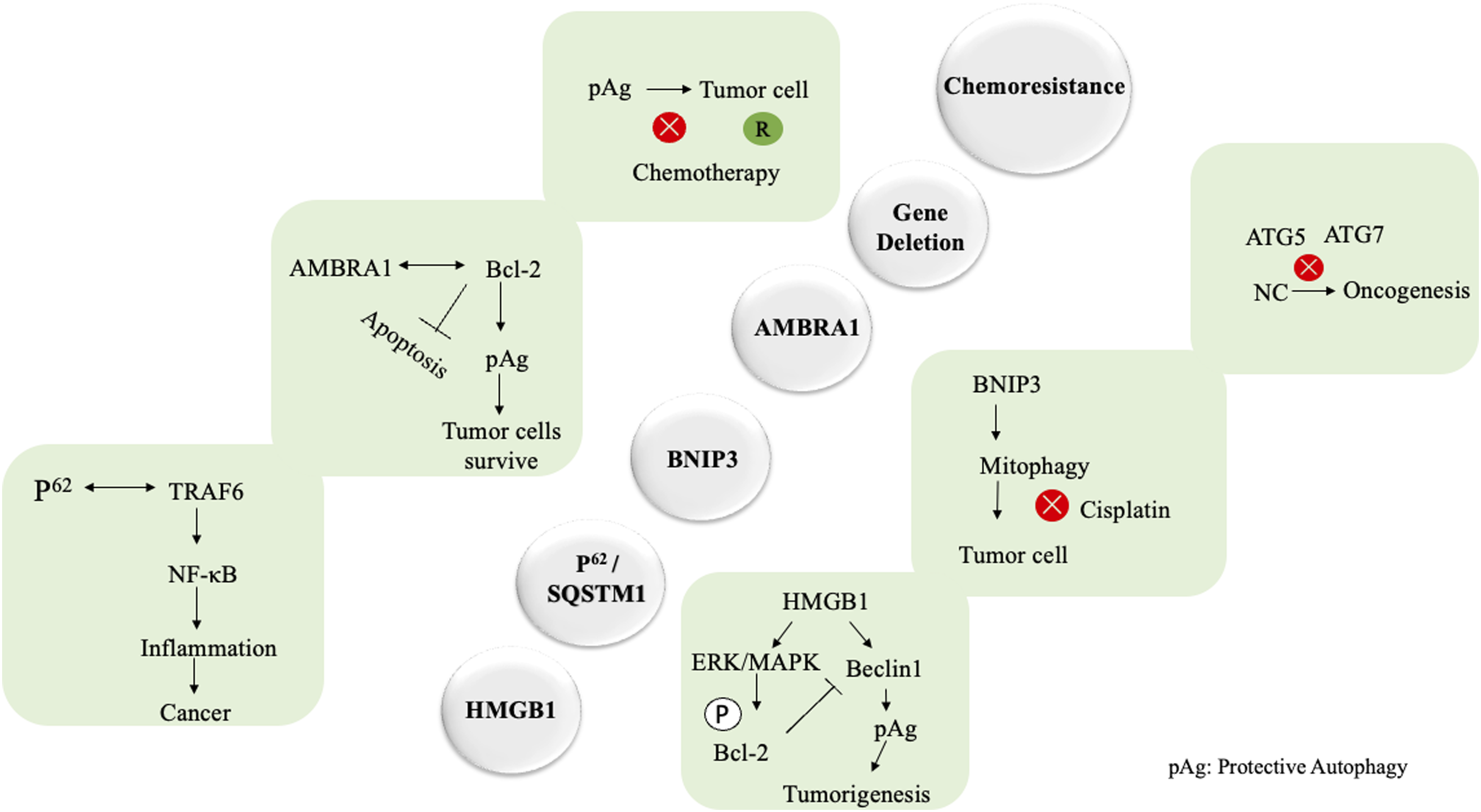
AMBRA1
AMBRA1 is a known positive regulator of autophagy. AMBRA1 plays a role in the early stage of autophagy by binding to Beclin-1 and facilitating Class III PI3K complex formation. 132 It has been proven that Beclin-1 dependent autophagy and apoptosis were regulated by a dynamic interaction between AMBRA1 and Bcl-2. 133 An in-vitro study on human fibroblasts and embryonic stem cells confirmed the key functionality of AMBRA1. 134 A study conducted on CRC cells demonstrated that the interaction between AMBRA1 and Beclin-1 facilitates autophagy. 134 Further data on in vitro and in vivo CRC cells indicated that autophagy activation contributes to the survival of cancer cells in TME. 135 Autophagy activation in CRC cells protects tumor cells from stress conditions and inhibits apoptotic cell death. AMBRA1 can regulate the balance between autophagy and apoptosis in cancer cells. 134 It has been observed that in response to apoptotic stimuli in CRC cells, AMBRA1 levels decreased. This observation indicates AMBRA1 can function as a negative regulator of apoptosis in CRC cells. 136 In summary, as a key regulator of autophagy, AMBRA1 also facilitates the pro-survival of CRC cell lines by inhibiting apoptotic cell death and enhancing stress relief mechanisms through autophagy.
P62/SQSTM1
The p62 is a prominent scaffold protein that provides substrate for autophagy, also known as sequestosome 1.20,84 The characterization of p62 protein is to recognize, bind, and transport ubiquitinated cytosolic materials for degradation. 137 In light of the upregulation of p62 protein, recognized as a potential marker of decreased autophagic machinery, implying p62 proteins as an oncogene. 137 In oxidative stress, p62 is upregulated, which facilitates the release of NRF2 from the KEAP1 protein. 20 Thereby, free NRF2 can transport to the nucleus to activate antioxidant defense genes, leading to tumorigenesis. 138 In several mouse models, it has been observed that autophagy deficiency results in overexpression of p62 and NRF2, which can promote tumor formation. 20 Another potential tumor-promoting mechanism accompanied by p62 is NF-κB-mediated inflammation. It has been proven in several studies that NF-κB is a potential inflammatory mediator 137 ; in autophagy-deficient cells, accumulated p62 protein can polyubiquitinate TRAF6. 139 This polyubiquitination of TRAF6 is needed for NF-κB activation. After activation, NF-κB is transported from the cytoplasm to the nucleus to increase IL-1 and IL-6 expression to facilitate inflammation. 137 A study demonstrated the downregulation of p62 through siRNA, inhibiting IL-1 and IL-6 expression. 137
BNIP3
BNIP3 is a known hypoxia-dependent protein that has a potential role in cell necrosis, apoptosis, and autophagy regulation through the mitochondrial pathway.33,140 Recently, BNIP3’s role in carcinogenesis was highlighted, as increased expression of BNIP3 was observed in primary prostate, laryngeal, gastric, glioma, cervical, melanoma, invasive breast, lung, hepatocellular carcinoma, and pancreatic ductal adenocarcinoma cancers.140,141 Alternatively, BNIP3 levels were repressed in normal cells to control normal cell growth. 141 Hypoxic conditions allow the induction of abundant BNIP3 through HIF-1α to facilitate cell proliferation and invasion. 141 In hypoxia, BNIP3 protects tumor cells from necrosis by nurturing autophagy. 142 Moreover, BNIP3-induced mitophagy leads to resistance to cisplatin in tumor cells. 143 Under hypoxic conditions, the reduction of hypoxia-induced cell death, retinoblastoma (Rb), a known tumor suppressor, attenuates BNIP3 activity. 144 Recently, several studies demonstrated HIF-1α-independent BNIP3 regulatory expression in different cancer cells. In particular, phosphorylated JNK attaches to the BNIP3 gene in the promoter region to facilitate upregulation of BNIP3 in hepatocellular carcinoma. 145 Additionally, in pancreatic cancer cells, overexpression of BNIP3 was reported through ERK (extracellular-signal-regulated kinase). 140 Overall, lower BNIP3 expression is observed in primary tumors, whereas elevated expression of BNIP3 is considered a marker of mature tumors. 141
HMGB1-Induced Autophagy
The HMGB1 is a nonhistone chromosomal housekeeping gene.146,147 This gene is highly expressed and widely distributed both internally and externally in cells. 147 Substantial research has identified the abnormal activity of HMGB1 in different cancer survival, including pancreatic, glioma, NSCLC, breast, colon cancers, and melanoma. 147 In a stressful condition like metabolic stress, oxidative stress, or genomic instability, HMGB1 can regulate autophagy, resulting in tumor cell survival. 148 It has been reported that HMGB1-induced autophagic activity depends on ROS. Under cellular stress, ROS production regulates HMGB1 translocation from the nucleus to the cytosol. 148 In the cytosol, autophagy activation depends on the disruption of the Beclin-1 and Bcl2 complex. HMGB1-induced autophagy is activated in response to phosphorylation of Bcl2, leading to PI3KC3 complex formation. 149 Another study revealed that Beclin-1 and Bcl2 disruption can be facilitated through activation of ERK/MAPK signaling, which also depends on HMGB1. 150 Overall, HMGB1-induced autophagy can be a potential target in different cancer therapies for better patient outcomes.
Autophagy in Cancer Metastasis: Orchestrating Dissemination and Cellular Fate
While the aspect of autophagy is increasingly critical in the metastatic cascade, recent evidence demonstrates that autophagy is central to every step in it, including local invasion, intravasation, survival in the circulation, extravasation, and colonization at distant organs. 151 Autophagy regulates cytoskeleton dynamics, epithelial-mesenchymal transition (EMT), and extra-cellular matrix (ECM) remodeling; thus, improving the migration and invasion abilities. 152 As an example, autophagy has been demonstrated to degrade adhesion molecules in cells such as E-cadherin, thus allowing EMT which is a requirement before detachment and spread of the cells. 153 Moreover, it is observed that autophagic activity plays a role in metabolic remodeling that maintains the energetically demanding cell motility and invasion. Detached tumor cells will face an issue of anoikis during circulation ie, a type of programmed cell death caused by loss of ECM attachment. Autophagy imparts resistance to anoikis through the sustenance of homeostasis and restriction of ROS build up. 154 Moreover, to endure oxidative and shear stress in the blood, circulating, tumor cells (CTCs) also employ autophagy.
However, autophagy also appears in context-specific functions at metastatic site. Compared to those with low autophagic flux, high levels of autophagic flux in some models favor dormancy-inducing the survival of the disseminated tumor cells (DTCs) in a quiescent state before they reactivate in new conditions. 155 On the other hand, autophagy in other situations sustains outgrowth through metabolic flexibility and evading immune surveillances. The precise mechanism of this effect is that the regulators, including BNIP3 and Beclin-1, regulate mitophagy to restraint mitochondrial reactive oxygen species to facilitate the migration and survival in the hypoxic environment that is characteristic of the pre-metastatic niche. 156 In addition, autophagy overlaps with pro-metastatic factor secretion such as matrix metalloproteinases (MMPs) and VEGF and cytokines that alter the ECM and bring in stromal assistance.
As shown in Figure 5, these processes together reveal how autophagy can promote EMT induction, ECM degradation, cytoskeletal reorganization, and secretome-mediated interactions with the stroma to facilitate invasion and intravasation. Moreover, autophagy promotes CTC survival in the circulation and immune escape through redox balance and inhibition of anoikis. High autophagic flux at the metastatic niche facilitates either dormancy or metastatic outgrowth in response to microenvironmental signals, with a dual role in the process of dissemination and colonization. Autophagy in EMT and the Metastatic Cascade. A Schematic Showing How Hypoxia/TGF-β Induce EMT (via SNAIL/TWIST/ZEB) and Recruit Autophagy Modules (Beclin-1, LC3-II, p62, BNIP3) to Enable E-cadherin Turnover, Cytoskeleton Remodeling, Anoikis Resistance, Immune Evasion, and a Pro-metastatic Secretome Across Invasion → Intravasation → CTC Survival → Extravasation → Colonization/Dormancy → Outgrowth
Interplay Between Autophagy (Ag) and Tumor Microenvironment (TME)
Tumor cells co-exist with diverse immune cells, stromal cells, extracellular matrix, and cancer-associated fibroblasts within a highly complex and heterogeneous ecosystem to form a Tumor Microenvironment (TME) structure. 157 It has been reported that 50%–60% of tumors thrive under hypoxic conditions known as TME factors, which contribute to therapy resistance, escape from immunosurveillance, and tumor cell survival. 158 Hypoxia is characterized by lower oxygen pressure, usually less than 5–10 mm Hg. 159 Thus, lower O2 pressure leads to the accumulation of stable HIF-1α in the cytoplasm to upregulate BNIP3 and BNIP3L, subsequently disrupt Beclin-1 and Bcl-2 association, and activate autophagy. 160 Activated autophagy will restrict tumor cell death in lower oxygen conditions. 159 Furthermore, hypoxic conditions within TME can restrict immune cells’ mediated tumor inhibition. It has been hypothesized that during intracellular trafficking, granzyme B is degraded through autophagosome-mediated degradation, as evidenced by the significant elevation of granzyme B levels upon Beclin-1 deletion in hypoxic tumor cells. 159
Research on TME has highlighted the critical function of immune components like dendritic cells (DCs), macrophages, and T and B lymphocytes in the surveillance and elimination of cancerous cells, ultimately determining the fate of tumor cell. 157 Effective immune cells, particularly T and NK cells, are key drivers of cancer cell clearance within the TME. 161 Further, it has been reported that autophagy in immune cells displays plasticity within the TME, acting either to hinder or enhance anti-tumor immune responses. Autophagy participates in various aspects of immune function, including the functional activation of T cells, B cells, DCs, and macrophages, along with the production of IgM and IgG.36,162 Conversely, in the context of anti-tumor immunity, autophagy can regulate immune cells in a negative manner, such as degrading cytotoxic granules (granzyme B, perforin, and connexin-43) derived from CD8+ T and NK cells, thereby inhibiting their ability to eliminate cancer cells. 163
Similarly, in melanoma cells, lysosomal proteolytic activity can impair CD8+ T cell function. 164 In addition, some studies show, the influence of autophagy in immunogenic cell death is through the regulation of DAMP levels. 165 Further, an in vivo study in autophagy-competent mice revealed that the deletion of ATG5 in CD8+ T cells can significantly restrain tumor growth, suggesting a context-dependent role for autophagy in CD8+ T cell-mediated anti-tumor immunity. 163 An immune-suppressive environment can be established by Treg in TME by producing inhibitory cytokines such as IL-10, IL-35, and TGF-β, to reduce the anti-tumor efficacy of T cells. 166 Another anti-tumor suppressive mechanism of Treg is to induce apoptosis via caspase in target immune cells. 167 Additionally, tumor recognition via antigen presentation is an effective anti-tumor immune response. 168 As mentioned earlier, autophagy is involved in DCs’ regulation, which eventually promotes antigen presentation and stimulates the CD8+ T cells’ response. 169
Conversely, autophagy can also facilitate tumor evasion from CD8+ T cells by degrading MHC-I on both pancreatic ductal adenocarcinoma (PDAC) and melanoma.170,171 In a PDAC mouse model, the presence of an autophagy inhibitor resulted in a significant improvement in MHC-I levels, leading to effective anti-tumor response by CD8+ T cells.
171
Some studies have shown that the activity of immune cells is limited by cytokine components in the TME; specifically TGF-β, which facilitates the reduction of the anti-tumor activity of NK cells by converting peripheral CD4+ T cells into Treg cells.
172
Conversely, the anti-tumor activity of NK cells is improved in breast cancer, treated with TGF-β inhibitors.
173
Further research on the immune-evasive mechanism of autophagy in TME is required to establish a novel therapy to improve clinical outcomes in cancer patients (Figure 6). Schematic Representation of the Interplay Between Autophagy and the Tumor Microenvironment (TME). Tumor Cells Interact With Immune Cells (CD8 + T Cells, NK Cells, Tregs, Dendritic Cells, Macrophages), Stroma, and Hypoxic Conditions that Induce Autophagy. Autophagy Mediates Immune Evasion by Degrading Cytotoxic Granules and MHC-I, Modulates Cytokines (TGF-β, IL-10), Promotes ECM Remodeling via MMPs, and Supports Tumor Survival, Dormancy, and Metastasis
Therapeutic Resistance Mediated by Autophagy
FDA-Approved Drug Resistance Mediated by Autophagy

Critical Challenges in Clinical Translation of Autophagy-Targeting Therapies
Although preclinical evidence on autophagy inhibition as a method to address therapeutic resistance triggers optimism, translation into clinical practice is not always successful and often discouraging. 202 Such a translational gap indicates the existence of some fundamental unresolved questions about the multifaceted and context-dependent nature of autophagy in cancer. The most significant contributory factor to the conflicting clinical phenotypes is that autophagy may act as a tumor suppressor or tumor driver, based on the cancer stage, type, and microenvironment messages. Early in tumorigenic stage, autophagy has the ability to halt tumor expansion via the elimination of malfunctioning organelles and the restriction of genomic instability. 48 But in the mature tumor, it can support the survival of cancer cells during metabolic stress, which aggravates therapeutic resistance. A number of clinical trials using autophagy inhibitor drugs including chloroquine (CQ) and hydroxychloroquine (HCQ) combined with chemotherapy or targeted therapy have yielded inconsistent results. 203 As an example, although some trials in glioblastoma and pancreatic cancer have reported improved progression-free survival, others have reported little or no benefit, with wide variation between different types of cancer. This has been hindered by methodological issues such as poor selection of patients, absence of validated biomarkers and inability to monitor autophagic flux with precision in patients. Additionally, the available inhibitors are not specific and may have off-target effects, which makes dosing and the safety profile difficult.
The etiology that other elements of autophagy dependency is heterogeneous in different cancers, is another important variable. Pancreatic ductal adenocarcinoma and Ras-mutant cancers are highly dependent on high basal autophagic flux and theoretically the most suitable to be targeted by autophagy inhibitors. 204 Nevertheless, even within the same cancers, a subpopulation of cells can vary in their sensitivity to contrasting results. In addition, the interaction of autophagy with other cell death processes such as apoptosis and necroptosis imply that even attempts at blocking it may actually have a paradoxical effect of stimulating tumor growth as cells become more malignant. 205 Additionally, preclinical models are typically not able to recapitulate the human tumor complex microenvironment of cancer. As an example, immune-evasive mechanisms of autophagy, including degradation of MHC-I and the cytotoxic granules, evince that autophagy inhibition may increase anti-tumor immunity in certain situations. 206 However, it is also possible that autophagy can be blocked systemically and affect the ability by immune cells to practice immune surveillance and antigen presentation. There is little understanding of this two-way influence.
Tumor Heterogeneity – Implications for Sampling and Monitoring
Heterogeneity of tumors is a patient and cancer type-specific challenge to utilization of autophagy biomarkers in cancer. Autophagy appears to be spatially heterogeneous in tumors, both activity and levels of activity can vastly differ in the hypoxic center, nutrient-deprived niches, and proliferative edges. In pancreatic cancer, hypoxic areas have an increased mitophagy, mediated through BNIP3, with a lower flux at the peripheral zones. 140 A single-core biopsy may be insufficient to provide an accurate picture of overall autophagic process in such tumors, possibly making use of inappropriate therapies. Along with spatial differences, temporal dissimilarities are an imperative factor. It is found that autophagic activity develops in the face of such selective pressures as chemotherapy, immunotherapy, or nutrient stress. 76 Initially low-flux tumors can develop resistance phenotypes through autophagy in the course of several weeks after initiating treatment. This plasticity indicates how tests at diagnosis are inadequate. 155 The longitudinal monitoring is needed to achieve reliable clinical guide by the autophagy biomarkers. The ability to detect exosomal LC3-II or autophagy-related microRNAs or circulating Beclin-1 have developed advances in non-invasive liquid biopsy to monitor these temporal changes.
The single-cell sequencing also underlines intra-tumoral heterogeneity such that, even in the same tumor, distinct autophagy signatures are observed across clonal populations. 207 This heterogeneity makes it difficult to establish universal thresholds of biomarkers and indicates that a contextual interpretation (tumor microenvironment, as well as evolutionary stage) is needed. Clinically, this would require multi-regions biopsies during baseline characterization and serial sampling during therapy to better define treatment strategies on an as-needed basis. The implication of this heterogeneity is enormous. Therapeutic protocols aimed at exploiting autophagy will necessarily have to be patient specific not only across cancer subtypes but also on a per stage per patient basis. 208 Dynamic heterogeneity supports arguably the need of adaptive therapeutic approaches wherein autophagy inhibitors are instituted, withdrawn, or combined on the basis of real-time biomarker monitoring. Incorporation of these strategies in the design of clinical trials will ensure that autophagy-directed therapies represent the biologic heterogeneity of tumors and maximize clinical efficacy.
Future Direction
The profound understanding of autophagic regulation, paradoxical activity, and resistance against chemotherapy opens a new question about the possible future strategies to overcome autophagically induced negative effects. As mentioned earlier, combination therapy has been experimented with in some clinical trials. The cytotoxic effects are one of the main concerns in combination therapies. We suggest the use of biomarkers not only in diagnosis but also as drug conjugates to ensure selective delivery to specific tumor cells, thereby increasing the drug efficacy
209
(Figure 7). Possible Strategy Through an Autophagy-specific Biomarker to Reduce Resistance and Ensure Normal Cell Survival in a Therapeutic Environment. Under Therapeutic Stress, Autophagy-specific Proteins’ Abnormal Upregulation can be Detected by Biomarkers. Autophagy Inhibitor Tagged With a Biomarker to Ensure Targeted Cell Identification, Clearance, and Safety of Normal Cells
Overview of Autophagy-specific Biomarkers in Different Cancer Cells

Therefore, overexpression of LC3-II indicates a higher level of autophagy activation to protect tumor cells from stress. Increased levels of LC3-II in patients provide a piece of valuable information to design a specific therapeutic approach for better clinical outcomes. In addition, LC3-II or p62 can conjugate with targeted drugs, which will identify abnormal biomarkers expressed in cells to deliver target drugs for better drug efficacy, while ensuring healthy cell survival. Overall, more in-depth research is required to find the potential autophagy-specific biomarkers for early tumor identification and evaluation of the treatment efficacy on tumor cells.
Clinical Implementation of Autophagy Biomarkers
Although the finding of biomarkers specific to autophagy (Beclin-1, LC3-II, ULK1, p62, and PTEN) suggests potential avenues in terms of early diagnosis and pharmaceutical intervention, the conversion of the given knowledge to clinical practice also requires tight validation and standardization. The development of a biomarker with clinical actionability requires strong analytical validity, clinical validity and clinical utility. There are now numerous autophagy biomarkers in preclinical or early-phase clinical development, but most have not been tested in large multi-center studies where they may demonstrate the consistency and accuracy of diagnosis.
As an example, it has been found that LC3-II overexpression has great prognostic importance in triple-negative breast cancer (TNBC) and hepatocellular carcinoma, where they are associated with negative patient outcome and with autophagic flux. 218 Nonetheless, there are no standardized cut-offs regarding the levels of LC3-II expression levels, thus preventing their reproducibility between laboratories. Immunohistochemistry (IHC) is the most common diagnostic tool still in use, however, variations in antibody production, sample fixation and scoring processes may affect the diagnosis accuracy. 219 It is also important to develop standard methods of sample collection, processing, and interpretation to incorporate LC3-II into the standard pathology initiatives.
Likewise, Beclin-1 was found to be involved in tumor suppressor biomarker status in breast, prostate and ovarian cancer cases where its lower expression was associated with higher stages of the disease and decreased survival rates. 81 Beclin-1, despite its potential, is subject to various regulating pathways, as well as it can be differentially expressed in the tumor microenvironment. This challenges the clinical interpretation of single-time-point measurements. Its utility may be enhanced by longitudinal follow-up and the creation of liquid biopsy methods, eg, levels of circulating tumor DNA or exosomal Beclin-1, to monitor response or disease progression.
The validation also depends on a biomarker and the type of cancer. As an example, the use of p62/SQSTM1 as an autophagy inhibitor is justified by the experimental evidence, and few clinical studies have been performed, which limit the utility in practice as there are no standardized thresholds and prospective clinical trials that show predictive performance. 220 Adding p62 measurement into multi-plex panels with known markers may improve both sensitivity and specificity, to paint a more complete picture of autophagic processes.
Regarding implementation, diagnostic laboratories should favor low cost of implementation, ease of integration, and challenges of reimbursement. To use biomarkers as a guide to therapeutic interventions, biomarkers need to be backed by companion diagnostics or FDA-approved assays as needed. The practice is especially applicable to new tactics that may pair biomarkers with drugs conjugates or targeting agents to increase tumor specificity and avoid the undesired byproducts of off-target interactions.
Clinical Integration Into Practice – Decision-Making Algorithm
The lack of an actionable framework for the manner in which oncologists can integrate autophagy-related biomarkers into their daily clinical practice has been identified as one of the factors inhibiting the clinical translation of these markers. 221 Candidates that have been reported to show potential include LC3, Beclin-1 and p62, but these are usually reported with descriptive or prognostic intention, rather than to work into a solid diagnostic pathway. To fill this gap, we present a streamlined algorithm of how and when to measure autophagic status, and how to use these results to inform a therapeutic plan. The algorithm would enter at the cancer diagnosis phase, hence the conventional histopathology and molecular profiling. Particular cancers that have been shown to be highly dependent on autophagy, including pancreatic ductal adenocarcinoma (PDAC), triple-negative breast cancer (TNBC) and hepatocellular carcinoma (HCC) are selected to be tested further.
Biomarker testing using IHC to assess LC3, Beclin-1 as well as p62 is recommended and can be supplemented with liquid biopsy in cases when this is possible. 222 These markers offer a reflection of autophagic flux that could enable clinicians to stratify patients into those with tumors that exhibit high or low autophagic activity. The combination therapies, such as combination of autophagy inhibitors and chemotherapy, radiotherapy or immunotherapy, could be used in patients with high levels of autophagic flux that will improve therapeutic efficacy by reducing treatment resistance. 223 In contrast, the patients with preserved or low levels of autophagy will then continue with standard treatment without incurring the extra risk of investigational inhibitors. Remarkably, longitudinal monitoring of autophagy during treatment by repeat biopsy or liquid biopsy is integrated within the algorithm. This step guarantees flexibility to adjust therapy on the fly in response to tumor mutation and resistance.
Figure 8 shows a simplified but clinically applicable clinical-pathway to incorporate the assessment of autophagy into oncology practice. It has strengths in its flexibility to be adapted by clinicians in many ways based on the type of cancer being treated, the availability of resources to treat it, and the patient-specific needs. The algorithm is not designed to substitute the expertise of oncologists, but rather a set scaffold to provide a more systematic way of integrating autophagy biomarkers in personalized medicine. This kind of orderly steerage is important in the conversion of bench-side discoveries into meaningful bedside tactics. The integration of autophagy assessment into a specific clinical pathway will increase diagnostic accuracy, maximize treatment assignment, and provide the platform to more normal clinical studies that could confirm autophagy-based therapies in a variety of cancer subsets. Evaluation of Autophagic Status in Therapeutic Decisions. A Clinical Decision-Making Algorithm Outlining when and How Autophagic Biomarkers (LC3, Beclin-1, p62) Should be Assessed in Oncology
Pharmacoeconomic Considerations
The translation of autophagy biomarkers into clinical oncology can no longer be justified as a matter of biological plausibility or theoretical clinical potential but must consider economic tractability. The health systems, especially those with low- and middle-income countries consider in their applications of new diagnostics and have to do a strident investigation of the cost-benefit ratio. The recent evidence in oncology reveals biomarker-driven selection of treatment paths is often cost effective in the long-term, such as use of HER2 testing in breast cancer and EGFR/ALK testing in lung cancer, both of which reduced inappropriate exposure to expensive drugs with superior outcomes. 224 The process of autophagy characterization requires expenses associated with tissues-based tests (IHC or immunoblotting), molecular tests (qPCR or NGS panels), and liquid biopsies (exosomal- or ctDNA-based tests). The per patient cost of an autophagy biomarker assay would most likely be cheaper than the cost of advanced genomic sequencing at least unless attached to other multi-omic platforms that are already present at the tertiary care centers. Although initial spending is not insignificant, this could help avoid high-cost agent administration to patients who would not be likely to respond based on autophagy-mediated resistance.
Economic modeling further indicates that even a small survival enhancement of 3 to 6 months as a result of optimizing the choice of therapies using biomarkers would make such tests cost effective in terms of saving expensive hospitalizations, administering fewer adverse events, and avoiding futile use of therapies with tens of thousands of dollars’ worth of price tags per treatment cycle. 225 In resource-poor healthcare systems, sequential implementation is possible, and another project might be first used in the cancers most likely to respond to autophagy inhibitors (eg, TNBC, PDAC, HCC), followed by integration into the greater oncology practice. The centralization of autophagy tests conducted in central laboratories, as well as the integration of autophagy testing into an existing NGS or IHC plan, can further reduce marginal costs. Notably, the pharmacoeconomic implications are more far reaching than the cost savings and include access equity. Biomarker based systems improve the inequities between patients treated in a biological or shotgun manner and those treated using biologically precise approaches. 226 To policymakers, the advent of autophagy biomarker testing is a two-pronged promise: clinical utility and cost-effectiveness. Although additional real-life cost-effectiveness research is mandatory, established economics principles strongly endorse the opinion that bio-markers of autophagy-directed interventions will be cost-effective and affordable even in constrained environments.
Regulatory Landscape for Autophagy Biomarkers
Regulatory approvals are essential to the translation of autophagy biomarkers to bedside, with the majority of such biomarkers most appropriately considered as companion diagnostics (CDx). 227 Both the Food and Drug Administration (FDA) of the U.S and the European Medicines Agency (EMA) have tough sets of frameworks that dictate the validation and approval of such biomarkers. These definitions require that CDx achieve the 3 key criteria of analytical validity, clinical validity, and clinical utility. Analytical validity refers to the ability of the biomarker assay to detect and measure autophagic status reproducibly and accurately (ie, autophagy level, on the basis of LC3-II puncta, Beclin-1, or p62 accumulation). Clinical validity means an association between the biomarker and therapeutic response or prognosis. Clinical utility describes an advantage of biomarker-driven.
In the United States, the FDA determines that CDx must be approved along with their corresponding therapeutic, usually through the Center of Devices and Radiological Health (CDRH). EGFR mutation testing, as an example, was simultaneously approved together with tyrosine kinase inhibitors. Considering applications to autophagy, such biomarker assays would probably require validation in phase III trials of autophagy-inhibiting agents (eg, using HCQ combinations). 228 In Europe, regulations are formed around In Vitro Diagnostic Regulation (IVDR, 2022) categorizing biomarker tests to select therapy as Class C high-risk diagnostics, which requires assessment by notified bodies, inter-laboratory reproducibility studies, and prospective clinical evaluation. 229
Comparative EMA and FDA Requirements for Autophagy Biomarkers as Companion Diagnostics

Priority Research Directions
In order to bring the potential discoveries regarding autophagy to the benefits of actual clinical applications, it is clear that research priorities need to be suddenly put in place. First, validations studies must be prospective with large multi-center studies on candidate biomarkers including LC3-II, Beclin-1, and p62. These examinations ought to employ standard immunostaining and identify a replicable cut-off point to relate to the accuracy in diagnosis and forecast across various groupings of patients. Second, longitudinal studies that evaluate the dynamic changes of disease patients in autophagic markers with regard to the response and building treatment resistance are needed. Examples of such studies might be liquid biopsy analysis of circulating tumor DNA, exosomes of autophagy proteins, or autophagy transcripts so that tumor behavior can be monitored non-invasively in real-time.
Moreover, integrating the mechanistic in-vivo studies and the functional genomics screens to characterize the tumor- and context-specific dependency on autophagy is required to fix the discrepancies observed across clinical use of autophagy-targeting drugs. Patient-derived xenograft models or organoid systems allow a more realistic representation of the tumor microenvironment and the discovery of the intersection between the control of autophagy and immune cell activity and metabolic reprogramming. In addition, the combination of autophagy inhibitors with other treatment options, including immune checkpoint blockade or metabolic inhibitors should be the priority of interest to be tested in phase I/II. Such trials should include strong biomarker endpoints in order to find responders and modify treatment strategy in response. Lastly, the field would progress satisfactorily with development of clinically accepted imaging or tracer that would detect and measure autophagic flux non-invasively in human cancer. New positron progression tomography (PET) indicators or autophagy indicators might empower clinicians to understand the efficacy of a drug in real time, which would allow them to create more customized treatment regimens.
Clinical Translation: Implications for Treatment Selection and Combinatorial Approaches
The linkage between mechanisms and practice in the field of cancer needs oncologists and researchers to convert the autophagy status into decisions concerning effective treatments. For example, tumors with high basal autophagic flux—such as pancreatic ductal adenocarcinoma and certain Ras-mutant cancers—may be more likely to benefit from combination regimens that include an autophagy inhibitor (eg, hydroxychloroquine) alongside chemotherapy or targeted therapy. 231 Biomarkers such as LC3-II or p62 may be utilized as pre-treatment patient stratification to order to indicate response to such combinations. 204 On the other hand, indiscriminate inhibition could end up to paradoxically drive progression in those cancers where autophagy is mainly a tumor suppressor at earlier phases (eg, some prostate or breast cancers with normal Beclin-1 levels). 232 Consequently, autophagy in particular should be used in patients selected by its stage-specific and tissue specific-role in order to prevent negative effects. This highlights the necessity of development of diagnostic assays which can responsibly measure the autophagic processes in biopsies of tumor or in blood in real time.
Practically, the combination therapy must be logically made to capitalize on the autophagy crosstalk with other pathways. As an example, the combination of autophagy inhibitors and the immune checkpoint inhibitors would promote antitumor immunity by blocking autophagic killing of cytotoxic granules and MHC-I, thereby creating successful immune surveillance. On the same note, metabolic regulation such that the metabolic blockade of glycolysis or oxidative phosphorylation used against tumor cells, in combination with autophagy blockade, could take advantage of the metabolic rigidity of the tumor during stress. In the design of clinical trials, the use of dynamic biomarker monitoring is vital to change dosing and prevent toxicity when it is not required. The response can also be assessed by real-time visualization of autophagic flux with PET radiotracers, or serial liquid biopsies, to monitor markers of exosomal autophagy. The combination strategies will become safer and more effective with clear algorithms concerning escalation, switching, and termination of therapy on the basis of these measures.
Integration with Personalized Medicine: A Multi-Omic Approach
Autophagy biomarker integration into personalized medicine roadmaps demands integrating them into multi-omic strategies that will fully characterize tumors. Precision oncology is becoming an increasingly important intersection of genomics, transcriptomics, proteomics, metabolomics and immunoprofiling to develop comprehensive therapeutic approaches. 233 The potential to complement these platforms lies with autophagy as a regulator of cell metabolism and cellular response to stress. In pancreatic cancer, an area of active research is defining the metabolic dependencies conferred by KRAS mutations, which are amplified by an increase in autophagic flux. 234 It is also plausible that integration of autophagy biomarkers like LC3-II-accumulation or Beclin-1 depletion with genomic analysis of KRAS and TP53 mutation status may serve to define patient subgroups with the greatest likelihood of positive responses to autophagy inhibitors. In TNBC, p62/ULK1 expression with immune gene transcriptomic signature (PD-L1, TIL density) can better predict outcomes with combined autophagy/immune checkpoint inhibition treatment. 235
This has been possible through next-generation sequencing (NGS) technologies that are ever more capable of multiplex biomarker panels that involve protein-based, RNA-based, and DNA-based characteristics. 236 In addition to multi-omic integration, computational models and artificial intelligence (AI) approaches have the potential in making clinical use out of autophagy data. The autophagy biomarker profiles, imaging characteristics, and clinical data can be absorbed by machine learning algorithms to produce predictive treatment-response models. Notably, this kind of systems can also represent temporal change, by predicting when a tumor could flip to the tumor-promoting state of autophagy, therefore informing dynamic therapy. 237 The incorporation of autophagy into multi-omic workflows also promises economic functionality through the use of shared platforms, diminished marginal costs of additional markers, and prevents siloed diagnostics. More importantly, it places autophagy in the evolving field of personalized medicine, where treatment is based not only on initial tumor phenotypic features but also on the current adaptive responses in the evolving tumor microenvironment.
Conclusion
The study of autophagy in cancer biology demonstrates its dual character towards being a tumor suppressor protein in initial stages of cancer development and a survival adaptation that drives cancer progression, metastasizing, and development of therapeutic resistance in advanced cancers. Although remarkable progress has been achieved towards unraveling the molecular regulators of autophagy and how it needs to talk to the tumor microenvironment, there is still a large number of gaps, especially in terms of making this knowledge clinically applicable. A potentially fruitful direction is the incorporation of the autophagy-specific biomarkers into the early detection, prognostic assessment, and stratification of treatment. These biomarkers have therapeutic value as correct identification of impaired autophagic processes may guide the process of choosing personalized therapies. Moreover, the conjugation of the biomarkers with the anticancer drugs may allow the highly specific delivery of the drug, potentially improving the efficiency of the treatment, whereas causing as little destruction to the healthy tissue as possible. Such a strategy could transcend the autophagic-dependent therapeutic resistance and preserve tissue integrity of normal tissues which is one of the most serious problems in contemporary oncology. Future biomarker-driven studies Precision oncology (also known as precision medicine) involves the ability to personalize approaches to cancer by considering risks and predicting outcomes.
Footnotes
Acknowledgements
We would like to sincerely thank Dr S. M. Bakhtiar UL Islam for his constant guidance and encouragement during the course of this work. His practical advice, careful review, and genuine interest in the topic helped us stay focused and thoughtful throughout the process. The manuscript benefited greatly from his involvement, and we are truly grateful for the time and attention he dedicated to it.
Author Contributions
The initial concept and structural framework of the review were developed by JS and SMBUI. JS and TA and were responsible for conducting the literature review, compiling and analysing the data, and drafting the manuscript. The writing process was overseen by JS, TA, MF, KR, and SMBUI, who also worked on refining and finalizing the manuscript. All authors reviewed and approved the final version for submission.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data Availability Statement
Data sharing not applicable to this article as no datasets were generated or analyzed during the current study.