
Abstract
Introduction
A few studies have examined whether the safety and efficacy of moderate hypofractionated post-prostatectomy radiotherapy (moderate HYPORT, also called MYSTERY) are equal to those of conventionally fractionated post-prostatectomy radiotherapy (COPORT) in patients with localized prostate cancer. Therefore, this study aims to compare the safety and efficacy of MYSTERY and COPORT in patients with postoperative prostate cancer.
Methods and analysis
This study is a prospective, single-center, open-label, randomized controlled clinical trial. Patients with localized prostate cancer will be randomly allocated to receive COPORT (66-74 Gy at 2 Gy per fraction) or MYSTERY (57.5-65 Gy at 2.5 Gy per fraction). The primary outcomes are radiotherapy-related gastrointestinal and genitourinary adverse events. Secondary outcomes include progression-free survival, quality of life, medical expenses, and overall survival.
Keywords
Strengths and Limitations of this Study
➢ A phase II study comparing MYSTERY and COPORT in patients with postoperative prostate cancer. ➢ To evaluate whether the safety and efficacy of the regimen of MYSTERY are equal to those of COPORT. ➢ The limitations of this study are that it is a single-center clinical trial, with a relatively small sample size.
Introduction
According to the latest World Cancer Report, in 2022 the incidence of prostate cancer accounted for 7.3% of all malignant tumors worldwide, ranking fourth in incidence and sixth in mortality. 1 The overall incidence of prostate cancer in China is lower than that in European and American populations, although its occurrence has grown rapidly over the past 20 years and now ranks first among male genitourinary (GU) malignant tumors. In 2022, approximately 47 500 deaths were attributed to prostate cancer in China, 2 with data indicating a persistent upward trend.
Localized prostate cancer is defined as prostate cancer confined within the prostate capsule, without evidence of lymph node or distant metastases. 3 International guidelines recommend radical prostatectomy (RP) as treatment for these patients. 4 For patients with postoperative risk factors for recurrence (pT3a, pT3b, pT4, positive margins, or N1), postoperative adjuvant radiotherapy is administered after recovery from postoperative urinary incontinence. Postoperative salvage radiotherapy is recommended for patients with prostate specific antigen (PSA) persistence or recurrence after RP,5,6 where PSA persistence is defined as undetectable PSA levels after RP with a subsequent detectable PSA level that increases to ≥0.1 ng/mL, failure of PSA to fall to undetectable levels, or PSA levels >0.1 ng/mL. 7 Some studies have shown that patients with high-risk pathological or biochemical factors after RP may benefit from pelvic adjuvant or salvage radiotherapy.8-10 Furthermore, the National Comprehensive Cancer Network (NCCN) guidelines 7 recommend conventionally fractionated post-prostatectomy radiotherapy (COPORT) (64 Gy–72 Gy at 1.8 Gy–2.0 Gy per fraction for a total of 7-8 weeks) ± androgen deprivation therapy for these high-risk patients.
Prostate cancer has a low α/β value (α/β < 2 Gy, generally 1.5 Gy), whereas surrounding normal tissues and organs, such as the bladder and rectum, have relatively high α/β values (3 Gy-10 Gy). Therefore, in theory, hypofractionated radiotherapy can provide better tumor control. 11 The 2018 American Society for Radiation Oncology, American Society of Clinical Oncology, and American Urological Association evidence-based guidelines classify hypofractionation into moderate- and ultra-hypofractionation. The moderate hypofractionation dose is 2.4 Gy–3.4 Gy, whereas ultrahypofractionation is defined as a single fractionation dose ≥5 Gy, and the single fractionation dose of 3.4 Gy–5 Gy does not have a clear definition because of a relatively little studied and little used intermediate range. 12 With the advancement of radiotherapy technology, intensity-modulated radiotherapy (IMRT) is widely used in clinical practice to effectively control the progression of primary lesions. As a result, recent NCCN guidelines have included hypofractionated radiotherapy (2.5 Gy–3 Gy per fraction for a total of 4-6 weeks) as a preferred option for prostate cancer. Compared with conventional fractionated radiotherapy, hypofractionated radiotherapy has a shorter treatment course, reduced medical costs, better compliance, and improved resource allocation, while also being more convenient for patients.
However, the literature on MYSTERY is limited, with most studies being retrospective. Several studies have reported a low incidence of toxicity with a single fractionation dose of 2.5 Gy, which is a feasible recommendation for dose segmentation.8,13-16 Moreover, there is a lack of evidence from high-grade clinical studies comparing the safety and effectiveness of COPORT and MYSTERY. Additionally, the optimal dose fractionation for MYSTERY has not yet been established. Therefore, this study aims to compare the safety and efficacy of MYSTERY at 2.5 Gy per fraction and COPORT at 2.0 Gy per fraction for prostate cancer treatment.
Methods and Analysis
Study Design
This prospective, single-center, two-arm, randomized controlled clinical trial is designed and supervised by investigators at Shanghai Changhai Hospital. The study protocol was developed and prepared in accordance with Standard Protocol Items: Recommendations for Interventional Trials (SPIRIT) guidelines.
17
Patients with histologically confirmed prostatic adenocarcinoma who have undergone RP will be enrolled in our study. A computer-generated randomization schedule will be used to randomly assign patients in a 1:1 ratio to either arm A (COPORT) or arm B (MYSTERY) (Figure 1). The random sequence will be generated by the Android app “RCT Assistant.” Flowchart of study treatments. MYSTERY: moderate hypofractionated post-prostatectomy radiotherapy; COPORT: conventionally fractionated post-prostatectomy radiotherapy; PB: prostate bed; PNI: prophylactic nodal irradiation.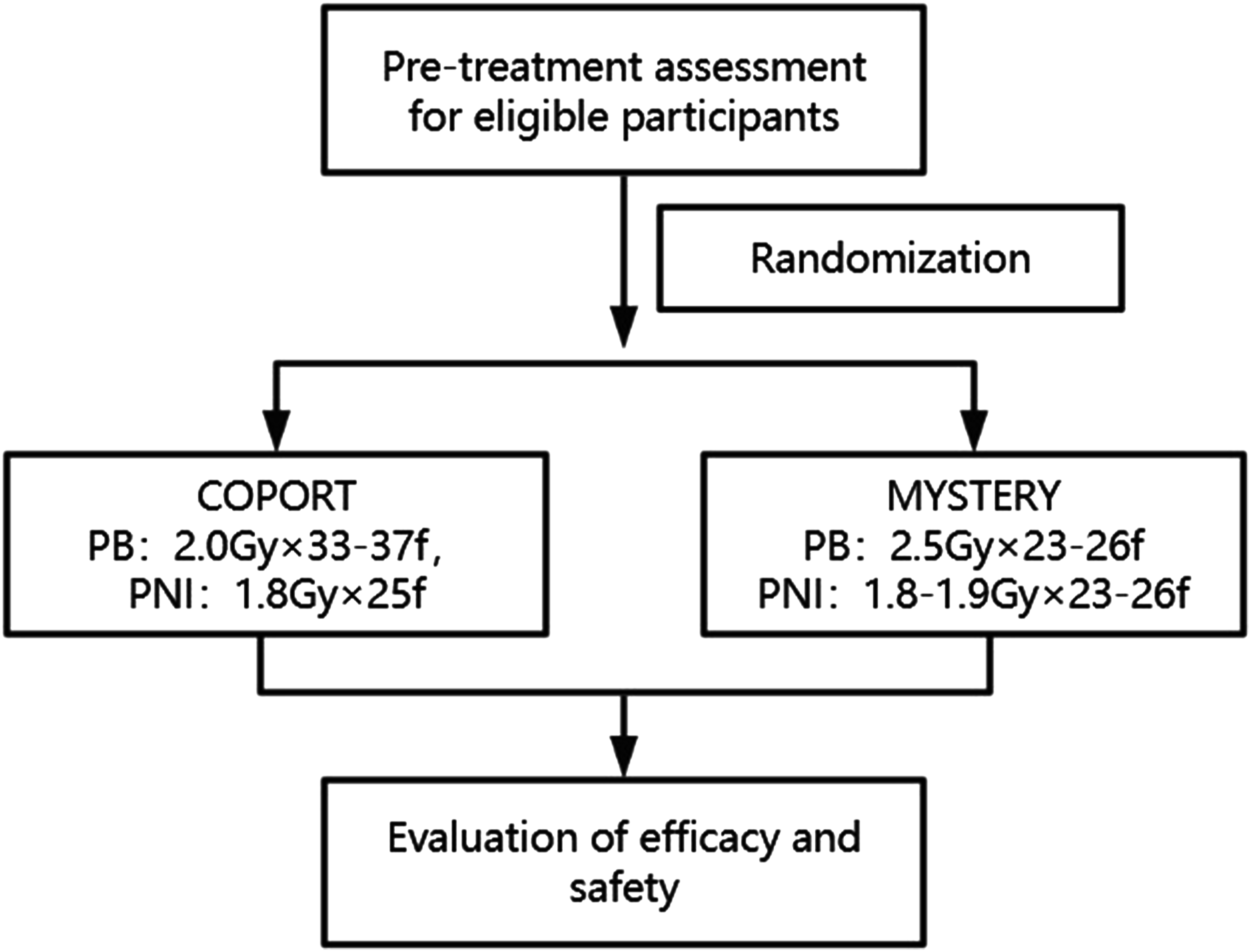
The Schematic Diagram for Data Collections and Assessment.

(1) Vital signs: blood pressure, heart rate, respiration, pulse, and abdominal examination.
(2) PSA: total PSA and free PSA.
(3) Blood routine: red blood cells, white blood cells, hemoglobin, platelets.
(4) Urine routine: red blood cells, PH, specific gravity, urine protein.
(5) Blood biochemistry: urea nitrogen, creatinine, Aspartate Aminotransferase, Alanine Aminotransferase, total bilirubin.
(6) Coagulation function: Prothrombin Time, Activated Partial Thromboplastin Time, Fibrinogen, Thrombin Time.
(7) Prostate mpMRI: multiparametric prostate MRI, including T2 weighted image, Diffusion-Weighted Imaging, Dynamic Contrast-Enhanced, and magnetic resonance Pope analysis.
(8) Pathological section analysis: it means the analysis of pathological specimens of prostate puncture.
(9) •: Required items; ○: Selected items (if disease progression).
Objectives
This clinical trial will compare the safety and efficacy of MYSTERY and COPORT in patients with postoperative prostate cancer.
Study Participants
Inclusion Criteria
➢ European Cooperative Oncology Group score ≤2. ➢ Patients with pathologically confirmed prostate cancer who underwent RP. ➢ Postoperative American Joint Committee on Cancer version 8 pathological staging of pT3a, pT3b, pT4, positive margins, or N1; or serum PSA ≥0.1 ng/mL 6 weeks after surgery; or serum PSA <0.1 ng/mL 6 weeks after surgery, with subsequent follow-up revealing 2 consecutive sustained PSA level increases (≥0.1 ng/mL) ➢ No signs of metastasis on clinical imaging, including magnetic resonance imaging (MRI), single-photon emission computed tomography (SPECT), and Ga-68 prostate-specific membrane antigen positron emission tomography / computed tomography (Ga-68 PSMA PET/CT). ➢ Expected survival >5 years. ➢ Patients who voluntarily consent to the experimental study protocol after being informed of the existing treatment options.
Exclusion Criteria
➢ Poor recovery of postoperative urinary continence. ➢ A previous history of pelvic or abdominal radiotherapy. ➢ Participation in other clinical trials that are mutually exclusive with the study intervention within 4 weeks prior to the start of the study. ➢ Patients with other malignancies and acute or chronic infections, including human immunodeficiency virus, hepatitis C virus, and syphilis. ➢ Patients who are unsuitable to participate in the clinical trial and may interfere with treatment, evaluation, and compliance, based on the judgement of the investigator. For example, patients with other serious systemic diseases, including severe respiratory, circulatory, neurological, mental, digestive, endocrine, immune, and urinary diseases, may be excluded. ➢ Patients with contraindications to radiotherapy. ➢ The inability to provide written informed consent.
Dropout Criteria
All patients who complete the informed consent and screening process and enroll in the study may be withdrawn from the clinical trial for the following reasons: ➢ The occurrence of grade 3/4 adverse events according to the Common Terminology Criteria for Adverse Events (CTCAE) v5.0. ➢ The trial is terminated for any reason (including poor compliance). ➢ Lost to follow-up. ➢ The patient requests to withdraw, for any reason.
If a patient withdraws from the trial, the investigator must fill out the cause of loss in the case report form, complete the assessment items and relevant tests, and complete the last condition record. For losses due to adverse reactions, the treatment method must be recorded in a case report form and the sponsor must be notified. Dropout cases will be entered into the efficacy and safety analyses based on different conditions. The effect and safety analysis of the case of dropouts will be analyzed according to the intention-to-treat method.
Randomization
The patients will be enrolled by the investigators and randomized in a 1:1 ratio between arm A (COPORT) and arm B (MYSTERY). A randomized list will be produced by the response technology provider, which ensures the random assignment of patients to randomization numbers after they are eligible. Each number will be linked to a treatment group.
The trial is open label. The study design and treatment allocation will not be masked to the patients, investigators, and all study members who conduct data collection because the outcomes of the clinical trial are objective.
Intervention
Radiation Therapy
Eligible participants will be randomized in a 1:1 allocation to receive either COPORT (arm A) or MYSTERY (arm B) (Figure 1). Treatment will be applied to the prostate bed, which includes the anastomosis, bladder neck, and rectal bladder space. Organs at risk include the rectum, bladder, femoral head, small intestine, colon, and bulbourethral glands.
Arm A: COPORT
Patients in arm A will receive COPORT over 7 weeks, which constitutes approximately 64 Gy–72 Gy in 32-36 daily fractions of 2 Gy to the prostate bed, in the absence of disease progression or unacceptable toxicity. For the entire pelvic region, 45 Gy in 25 daily fractions will be administered.
Arm B: MYSTERY
Volumetric modulated arc therapy is used for MYSTERY. Patients in arm B will receive MYSTERY over 5 weeks, which constitutes approximately 57.5 Gy–65 Gy in 23-26 daily fractions of 2.5 Gy to the prostate bed, in the absence of disease progression or unacceptable toxicity. For the entire pelvic region, 41.4 Gy–49.4 Gy in 23-26 daily fractions of 1.8 Gy–1.9 Gy will be administered.
Lymph Node Irradiation
Pelvic lymph node irradiation is recommended for patients with pN1 disease. For high-risk patients without adequate intraoperative lymph node dissection, pelvic lymph node irradiation should be considered. The irradiated areas include the total, external, and internal iliac, obturator, and S1-3 presacral lymph nodes.
Treatment in Cases of Progression
If treatment is unsuccessful and disease progression occurs, remedial treatment options will be discussed based on the patient’s condition; these options may include endocrine therapy, chemotherapy, stereotactic body radiation therapy, and other treatments recommended by the NCCN guidelines.
Date Collection
Table 1 shows a schematic diagram of the data collection and efficacy and safety evaluation. The pretreatment and follow-up data of all subjects will be evaluated by physicians and then re-examined by researchers not involved in the study to ensure accuracy and completeness.
All patient information will be kept strictly confidential. For example, each patient will be assigned a unique ID so that the analysis results can be deidentified. If required, the ethics committee and authorized researchers may retrieve the treatment and follow-up data from the database.
Follow-Up
Serum PSA and testosterone levels will be evaluated monthly. After completion of the study treatment, patients will be followed up once a month for the first 3 months, and then once every 3 months for at least 5 years. If patients show biochemical progression, MRI, SPECT, and Ga-68 PSMA PET/CT will be performed for further evaluation.
Outcomes and Measurements
The primary endpoints are radiotherapy-related gastrointestinal (GI) and GU adverse events. Toxicity will be assessed using CTCAE v5.0. Secondary endpoints include progression-free survival (PFS), QoL, medical expenses, and overall survival (OS). PFS includes biochemical PFS (bPFS) and radiological PFS (rPFS). QoL will be evaluated using the Expanded Prostate Cancer Index Composite Instrument (EPIC-26) and Physical Activity Rank Scale-3 (ARS-3).18,19
Determination of Sample Size
Assuming that the proportion of grade ≥2 GI and GU adverse events in the control group is 52%, 20 statistical analysis according to non-inferiority test requirements, show that at least 193 patients per arm (386 patients in total) are required to achieve 80% power, with an α of 0.025. Allowing for a 10% loss to follow-up, the total sample size should be 428.
Data Management and Monitoring
Our database will store patient data, including basic characteristics, medical histories, and clinical and laboratory examinations results. The accuracy of data entry will be verified by two administrators, and the Ethics Committee of Shanghai Changhai Hospital will be responsible for data monitoring.
Statistical Analysis
Statistical analyses will be performed using the SPSS software (IBM, Armonk, NY, USA). For the two study groups, normally distributed continuous data will be described as mean ± standard deviation, and a two-sample t test will be used for comparing the two groups. Continuous data with nonnormal distribution will be expressed as median (range), and the Wilcoxon test will be used for comparing the two groups. Categorical data will be expressed as n (%) and compared using the chi-square or Fisher’s exact tests. The PFS, bPFS, rPFS and OS of the two groups will be estimated using the Kaplan-Meier method and compared using the log-rank test. Statistical significance will be defined as P values <0.05.
For the non-inferiority test (to test whether the adverse reaction rate of the experimental group is no worse than that of the control group), the full data set will be selected for analysis to avoid overestimating the results. Data from all patients who received the randomized treatment will be analyzed based on the full-set principle. For loss to follow-up data, the intention-to-treat method will be used, where all participants’ data will be included to calculate efficacy, the experimental group will be regarded as invalid, the control group as effective, and the comparison will be repeated.
Patient and Public Involvement
Patients or the public were not and will not be involved in the design, conduct, reporting, or dissemination of the research.
Ethics and Dissemination
This study was approved by the Ethics Committee of Shanghai Changhai Hospital (CHEC2024-048) on January 29, 2024, and registered at https://ClinicalTrials.gov (NCT06325995). All eligible patients will be informed by their physicians about the details of the procedures, benefits, and risks of MYSTERY and COPORT. Patients will voluntarily decide whether to participate in the study. As the outcomes of the clinical trial are objective, treatment allocation will not be masked to the patients, investigators, or study members. However, the process of randomization will be performed by researchers who are not involved in the study. Patients may withdraw from the study at any time, for any reason. Physicians will record all adverse events promptly. The findings of this study will be disseminated in peer-reviewed scientific journals and at relevant conferences.
Discussion
For high-risk patients who have undergone RP, postoperative adjuvant radiotherapy may improve their prognosis. 21 In 2020, Chin et al. 11 reported the 10-year follow-up data of 112 patients who received HYPORT; they showed promising results, with an OS of 90% and 75% and cancer-specific mortality of 5% and 11% at 5 and 10 years, respectively. Furthermore, HYPORT has a good tumor control rate and low incidence of acute toxicity. In particular, the acute lower GI toxicity rate of IMRT is lower than that of three-dimensional conformal radiotherapy (P = 0.14). 22
Some studies have explored the optimal split and maximum tolerated dose of HYPORT. In 2013, Ippolito et al 16 conducted a HYPORT dose-escalation study and concluded that a dose of 2.5 Gy per fraction was safe. In their study, four groups had prescription doses of 2.27 Gy, 2.39 Gy, 2.45 Gy and 2.50 Gy per fraction in 25 fractions. The rates of grade 2 acute GI toxicity in the four groups were 4%, 4%, 8%, and 4%, respectively, and those of grade 2 acute GU toxicity were 12%, 12%, 8%, and 4%, respectively; no grade ≥3 acute toxicities were reported. In 2022, Patel et al. 23 conducted a study on 15 patients using different dose levels and proposed that the maximum tolerated dose of postoperative large fractionation radiotherapy was 44.2 Gy in 10 fractions. The three dose levels were 56.4 Gy in 20 fractions, 51.2 Gy in 15 fractions, and 44.2 Gy in 10 fractions. The study also reported that the incidence of grade 2 and 3 acute toxicity was 60% and 6.6%, respectively. Grade 2 late GI and GU toxicities were 20% and 20%, respectively. Grade 3 late GU toxicity was 13.3%, and no grade ≥4 acute or late toxicities were observed. Although these two studies proposed recommended and tolerated doses for HYPORT, the optimal dose remains to be established. Furthermore, consensus regarding the optimal regimen has not yet been reached. Thus, the study compared MYSTERY and COPORT in patients with postoperative prostate cancer. But the limitations are that it is a single-center clinical trial, with a relatively small sample size. If the results are that the safety and efficacy of the regimen of MYSTERY are equal to those of COPORT, further studies will be conducted. Consequently, an appropriate dose-segmentation scheme for MYSTERY is still being explored.
Conclusion
In conclusion, HYPORT has been shown to have good tumor control and a low incidence of acute toxicity; moreover, it has the potential to shorten treatment cycles, reduce health care costs, increase patient compliance with improved convenience, and improve the resource allocation of treatment centers. With the development of an increasing number of prospective randomized controlled trials, the safety and effectiveness of HYPORT will be further verified.
Footnotes
Acknowledgments
We warmly thank Dr Ada Xiong from CNNC Accuray (Tianjin) Medical Technology Co, Ltd For her help in the professionally stylistic ameliorations of the English language.
Author Contributions
Study conception: H.J.Z. Initial Study design: Y.Y.L. and X.Z.Z. Revision of study design and protocol: H.J.Z, X.Z.Z., Y.Y.L., W.W.Z. Study coordination: All authors. Drafting the manuscript: Y.Y.L. X.Z.Z. and W.W.Z. All authors read and approved the final manuscript.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Shanghai Top Priority Research Center Construction Program (2022ZZ01011) and The construction of the First Affiliated Hospital of Naval Medical University “the 14th five-year plan” program of discipline cluster (center) of urogenital system disease diagnosis and treatment.
Trial registration number
Clinicaltrials.gov identifier: NCT06325995; Pre-results.