
Abstract
Introduction
Colorectal cancer remains the second cause of cancer mortality and the third most commonly diagnosed cancer worldwide. 1 The global estimated incidence of colorectal cancer in 2020 was 1.9 million cases and is anticipated to increase to 3.2 million cases in 2040 due to population growth and lifestyle changes. 1 Wide variations in colorectal cancer incidence have been reported across the global regions with the increase attributed to economic growth, westernization of diet, and lifestyle patterns. 2 Migrants and epidemiological studies conducted over some time have supported the varying incidence of colorectal cancer trends due to lifestyle patterns.3,4 However, unchanging incidence have been recorded in high-income countries and the trend was attributed to effective screening and early detection, while incidence in low-and middle-income countries continues to rise.3,5 The increase in colorectal cancer risk has also been strongly linked to environmental factors including red/processed meat consumption, alcohol, tobacco, sedentary lifestyle, and obesity. 6
The term “colorectal cancer” describes cancer that originates in the digestive system’s colon or rectum. The colon, which is composed of the left colon (left one-third of the transverse colon, descending colon, and sigmoid) and the right colon (caecum, ascending colon, and right two-thirds of the transverse colon), with the rectum joining the colon, makes up the large intestine (large bowel). In the large intestine, the rectum holds stool until it is eliminated, whereas the colon, which is the longest and first portion, absorbs water and nutrients from food. Colorectal cancer arises from the uncontrollable proliferation of the inner layer of glandular epithelial cells in the colon or rectum, which leads to the formation of adenomatous polyps and ultimately adenocarcinoma. 7 The pathways to the development of colorectal cancer from normal epithelial cells are heterogeneous with the accumulation of genetic/epigenetic mutations and local inflammation alterations. 8 The morphology of these cancer cells comprises epithelial hyperplasia, atypical hyperplasia, adenoma development, carcinoma in situ, and invasive carcinoma. The classical (suppressor), and alternative (mutator) pathways are the 2 carcinogenesis models of colorectal cancer. Colorectal carcinomas can be categorized as sporadic, hereditary, or familial depending on mutation origin. Approximately 70% of all colorectal cancer cases are sporadic and develop via the adenoma-carcinoma pathway as a result of a particular series of mutations. 9
The adenoma-carcinoma pathway, which primarily adheres to Knudson’s classic two-hit hypothesis, is responsible for the development of sporadic colorectal cancer. The first gene to mutate via the Wnt signaling pathway is the gatekeeping gene, Adenomatous polyposis coli (APC), which results in the hyper-proliferation of epithelial cells. Ring Finger Protein 43 (RNF43), a regulatory protein of the Wnt signaling pathway, and Catenin Beta (CTNNB1)- a gene that encodes beta-catenin are 2 more genes that have been altered along this pathway. Next, through the MAPK signaling pathway, mutations in Kristen Rat Sarcoma Viral Oncogene homolog (KRAS) and other RAS family genes, such as BRAF and NRAS, cause cell cycle progression and encourage the production of bigger adenoma polyps and precancerous polyps. Larger precancerous polyps and the formation of adenocarcinoma tumors are the result of suppressing cell apoptosis and increasing cell proliferation through the loss of Mothers Against Decapentaplegic Homolog 4 (SMAD4) and Loss of Heterozygosity 18q (LOH 18q) genes via the TGF-β and PI3K signaling pathways. Several other genes have been altered along these pathways, including phosphatidylinositol 3-kinase catalytic subunit alpha (PI3KCA), transforming growth factor-beta (TGF-β) receptor type 2 (TGFBR2), cell division control protein 4 (CDC4), and phosphatase and tensin homolog (PTEN). Later in carcinoma, mutations in BCL2-associated X (BAX) and tumor protein 53 (TP53) are acquired. These mutations further inhibit cell death by activating the p53 signaling pathway
10
(Figure 1). The adenoma-carcinoma model describing tumor progression steps from normal epithelial to cancer stage. Mutation in adenomatous polyposis coli (APC) gene is the first gene to be mutated and through the Wnt signaling pathway, it leads to hyper-proliferation of the epithelial cells. Next, mutations in Kristen Rat Sarcoma Viral Oncogene homolog (KRAS) cause cell cycle progression through the MAPK signaling pathway to promote the formation of larger adenoma polyps and pre-cancerous polyps. The loss of Mothers Against Decapentaplegic Homolog 4 (SMAD4), along with Loss of heterozygosity 18q (LOH 18q) via the TGF-β and PI3K along with mutations in tumor protein 53 (TP53) via the TP53 signaling pathway are acquired later in adenocarcinoma stages. These mutations further suppress cell death and encourage cell proliferation and are reported as bad prognoses for colorectal cancer diagnosis.
Risk factors that influence the development of these mutations include gut microbiome, dietary composition, lifestyle patterns, and host immunity, and these factors independently or together cause somatic mutations which determine the rate of progression and aggressiveness of the tumor.6,11 The signs and symptoms of colorectal cancer depend on the location of the tumor with classic symptoms such as constipation, blood in stool, change in bowel habits, loss of weight, and tenesmus. However, about 50% of people do not experience any symptoms, and this poses a challenge for early diagnosis, critical in disease management. 11
Molecular Pathways Involved in Colorectal Cancer Development
Generally, the molecular alterations in colorectal cancer can be categorized into 2 main kinds: mutations that result in new/increased functions of oncogenes and mutations that result in loss of function of tumor-suppressor genes. 12 It is worth noting that most oncogenic and tumor-suppressor mutations in colorectal cancer are somatic mutations.7,12 Tumor development in colorectal cancer emerges through several processes that involve genetic alterations including; chromosomal instability (CIN), microsatellite instability (MSI), CpG island methylator phenotype (CIMP), and epigenetic alterations including DNA methylation, histone aberrations and changes in miRNAs expression. 13
Chromosomal instability (CIN) is the most prevalent type of genomic instability found in about 85% of all colorectal cancer cases and is defined as a marked increase in the gain or loss of large or whole chromosomes. 14 The activation of oncogenes (KRAS and BRAF), inactivation of tumor suppressor genes (APC and TP53), and loss of heterozygosity for the long arm of chromosome 18 (18q LOH) are characteristics of chromosomal instability in colorectal cancer, which in turn promotes the carcinogenesis of the cancer.15,16
Microsatellite instability (MSI) is another distinguishing feature of malignant cells, which is characterized by the presence of at least 30% unstable loci in a panel of 5-10 loci made up of mono- and di-nucleotide tracts chosen at a consensus meeting held by the National Cancer Institute. 17 Lynch syndrome is typified by MSI, which is present in >95% of HNPCC patients but only 15% of sporadic colorectal cancer cases. 14 The inactivity of the DNA mismatch repair (MMR) mechanism results in a 100-fold increase in the mutation rates of colonic mucosa cells, which is the cause of MSI.
CpG island methylator phenotype (CIMP) is a distinct mechanism via which colorectal cancer advances, marked by an extremely high frequency of methylation at the promoter region of genes. 18 About 50% of human gene promoter regions contain CpG-islands, which are 200-500 base pair lengths of nucleotides. These segments are typically richer in the dinucleotide sequence that consists of cytosine and guanine and are known as CpG-residues. Due to the significant role CpG islands play in gene regulation, methylation of the CpG islands in promoter regions silences the corresponding genes. 18 The number of methylation markers detected in the colorectal tumor tissue can determine the CIMP status, which can be divided into 2 groups: CIMP-positive or CIMP-negative, or into 3 groups: CIMP-high, CIMP-low, and CIMP-negative. 19 In 20%-30% of all colorectal cancer patients, both MSI and CIN, widespread hypermethylation of the promoter regions of numerous tumor suppressor genes leads to epigenetic silencing. 20 Almost all signaling pathways and cell regulatory mechanisms can be impacted by changes in methylation patterns. A gradual accumulation of these genetic alterations results in normal mucosa switching to benign adenoma, then to severe dysplasia, and finally to adenocarcinoma.
Epigenetics and Colorectal Cancer
Epigenetics involves modifications to DNA that generate changes that turn genes on or off, without changing the DNA sequence, and these changes could be due to age and/or exposure to environmental factors. 21 These changes, referred to as the epigenome, affect gene expression patterns and cellular activity without changing the genetic sequence. The changes include modifications to DNA or histone proteins as well as the regulation of non-coding RNAs, and have been indicated to play a significant role in colorectal cancer initiation and progression. 21
Although, epigenetics is important in normal development and differentiation, some epigenetic events including hyper/hypo DNA methylation, post-translational modifications of histones and chromatin structure and changes in microRNAs expression may result in either activation of oncogenes or deactivation of tumor suppressor genes. 22 It is important to note that DNA methylation is one of the well-reported epigenetic events. The interaction of dietary intake and alcohol consumption as well as microbial metabolites within host cells have been strongly associated with the pathogenesis of various cancers including colorectal cancer via aberrant DNA methylation in host cells. 23 In this narrative review, we discuss how microbiome dysbiosis, microbial metabolites, diet, and alcohol consumption affect epigenetic events in the development of colorectal cancer.
Microbiome Dysbiosis in Colorectal Cancer
The human gut houses several microbial communities involved in the structural, metabolic, and protection of the gut. 24 The gut microbiome participates in important processes such as food digestion and nutrient absorption; the synthesis of several enzymes and vitamins including folate; and the production of short-chain fatty acids (SCFAs). Acetate, propionate, and butyrate are fermentation by-products of gut microbiota that give energy to the epithelial cells and ensure gut health. The human gut microbiome is dominated by 4 bacteria phyla: Firmicutes, Bacteroidetes, Actinobacteria, and Proteobacteria while the minority bacteria phyla are the Fusobacteria and Verrucomicrobia. 25 Microbial dysbiosis is generally characterized by a reduction in commensal bacteria, an increase in opportunistic/potentially harmful bacteria, or a decrease in bacteria diversity. 26
The composition of the microbiome varies greatly from person to person, although in healthy adults, it is generally stable throughout time and primarily determined by genetics. A significant difference in the gut microbiome is seen in the gut of healthy individuals compared to diseased individuals. 27 Numerous environmental factors, such as diet, consumption of alcohol, chemical exposure, and the use of antibiotics and drugs, contribute to these variances. 28
A plethora of research conducted in recent years has established a direct link between changes in the gut microbiota and the development of colorectal cancer.29-32 A recent meta-analysis conducted on microbiota revealed a higher proportion of Peptostreptococcus stomatis, Gemella morbillorum, Bacteroides fragilis, Parvimonas spp., Fusobacterium nucleatum, Solobacterium moorei, and Clostridium symbiosum with lower proportions of Bifidobacterium, Eubacteria, Ruminococcus and Lachnospira in colorectal cancer patients than healthy controls. 33 In a study, intestinal mucosa and fecal samples from patients with colorectal cancer were found to show less richness and diversity of bacteria than samples from healthy individuals.34,35 In another report, healthy tissues surrounding colorectal cancer tissues had an overabundance of Firmicutes and Fusobacterium, and less of Proteobacterium. 29 The same study discovered a greater number of species of which Firmicutes, Bacteroidetes, and Proteobacteria were over 98% of the phylum detected in malignant tissues as opposed to healthy tissues. Remarkably, Hibberd et al revealed that the microbiota in biopsy samples from different colorectal cancer patients was less diverse than that obtained from healthy individuals. 36 This implies that the microbiota’s composition changes greatly in healthy persons and remains relatively stable in tumors, possibly impacting the development and course of colorectal cancer.
It has been demonstrated that the microbial community composition in patients with advanced adenoma is significantly different from patients with carcinoma, indicating a potential role for the gut microbiota in tumor growth. 37 Stool samples from patients with typical adenomatous polyps showed a decrease in species richness when compared to controls who had no polyps; however, there was no difference in overall composition or diversity between patients with hyperplastic polyps or sessile serrated adenoma. 38 Some bacterial species have been linked to the development of colorectal cancer. In particular, Blautia, Clostridium, Bifidobacterium, and Roseburia have been observed to be significantly reduced in colorectal cancer patients, whereas Fusobacterium nucleatum, Campylobacter, Escherichia coli, Peptostreptococus, Enterococcus faecalis, Shigella, and Streptococcus gallolyticus have been significantly increased. 37 These changes could lead to an increase in pro-inflammatory opportunistic infections and a decrease in butyrate-producing bacteria, which would upset intestinal homeostasis and perhaps lead to tumor formation. Fusobacterium spp. have thus also been shown to be more prevalent globally in adenomas than in adjacent normal tissues, suggesting a possible link between the bacteria and early dysbiosis indicators in adenomas. 39 The mechanism via which microbiota influences colorectal cancer development is by generating microbial metabolites that interact with the host’s immune and metabolic systems and release genotoxic virulence factors that affect genetic and epigenetic regulations.40,41
Pathogenic Bacteria and Colorectal Cancer
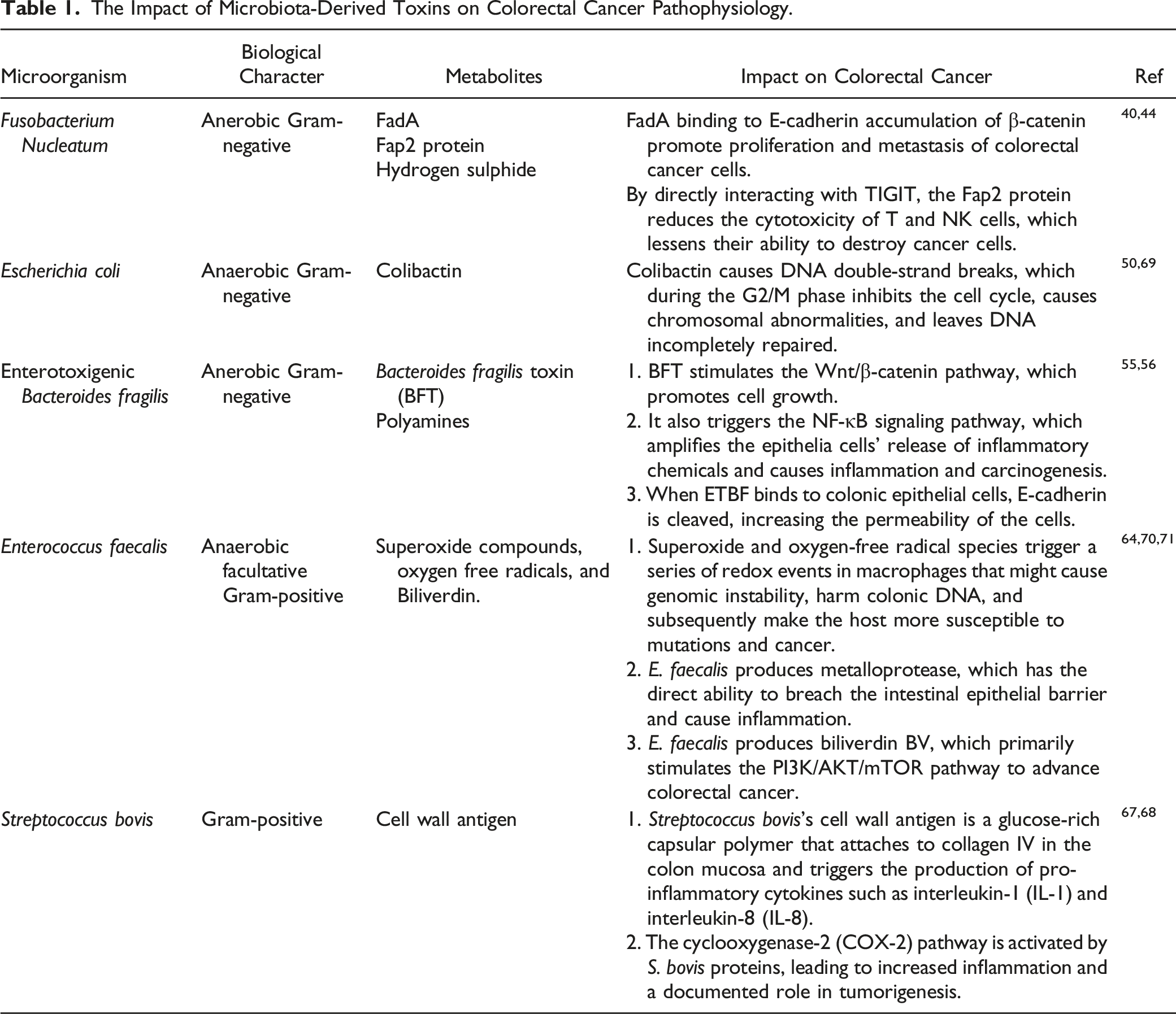
The development of colorectal cancer may be related to the presence of specific pathogenic bacterial species in the gut in addition to changes in the microbiota. A multitude of unique pathogens, including Fusobacterium nucleatum (F. nucleatum), Escherichia coli (E. coli), Bacteroides fragilis (B. fragilis), Enterococcus faecalis (E. faecalis), Streptococcus bovis (S. bovis), have been linked to the development of colorectal cancer through several different mechanisms including inciting inflammation, attaching to host epithelial cells, and secreting toxins40,41 (Figure 2). The interaction of microbiota-derived toxins on epithelial cell function and colorectal cancer pathophysiology.
Fusobacterium Nucleatum and Colorectal Cancer
Fusobacterium nucleatum (F. nucleatum) is an anaerobic gram-negative bacterium that thrives in the human oral cavity, gastrointestinal tract, and other areas. Elevated tissue concentrations of F. nucleatum have been associated with the involvement of lymph nodes and advanced stages of colorectal cancer. 39 One way F. nucleatum influences the development of colorectal cancer is by producing hydrogen sulfide, which binds to E-cadherin on the cell surface and enters epithelial cells through its surface virulence factor, Fusobacterium adhesion protein A (FadA). 42
It has been demonstrated that this leads to the production of proinflammatory cytokines, which support the growth of tumors in a microenvironment. 42 Although E-cadherin is normally a tumor suppressor, binding to FadA impairs its function. Thus, F. nucleatum is known as an initial driver in colorectal cancer pathogenesis and promotes metastatic colorectal cancer. 43 Moreover, the interaction between E-cadherin and FadA activates β-catenin signaling, which starts inflammatory and pro-oncogenic processes that lead to the development of cancer. 40 Fusobacterium autotransporter protein 2 (Fap2) is another adhesin that is derived from F. nucleatum. It can bind to the T cell immune receptor with Ig and ITIM domains (TIGIT), an inhibitory immune receptor, and reduce the activity of NK and T cells as well as promote immune cell apoptosis. 44
Escherichia coli and Colorectal Cancer
Escherichia coli, an aero-anaerobic gram-negative bacterium, is the predominant species in the normal gut microbiota; yet, some strains of this bacteria have been linked to several intestinal and extra-intestinal illnesses. E. Coli strains can be classified into 4 main categories based on phylogenetic analyses: A, B1, B2, and D. 45 Most commensal strains belong to group A, while virulent extra-intestinal strains primarily belong to group B2 and, to a lesser extent, group D. 46 Although E. coli is a common gut bacterium that is recovered from both colorectal cancer patients and healthy controls, yet, colorectal cancer patients have more pathogenic strains of E. coli than healthy persons.47,48 Aside from other cyclomodulins produced by E. coli such as cytolethal distending toxins and cytotoxic necrotizing factors, these pathogenic strains also produce colibactin, which when in contact with mammalian cells, is capable of inducing a specific cytopathic effect known as megalocytosis. 49
Genes responsible for producing colibactin are located on a particular genomic island known as the polyketide synthetase (pks) island, which is mostly found in the E. coli B2 phylogenetic group and is responsible for colibactin expression.49,50 Colibactin is a non-ribosomal peptide synthetase that causes DNA double-strand breaks (DSBs), which during the G2/M phase inhibits the cell cycle, causes chromosomal abnormalities, and leaves DNA incompletely repaired. 50 Colibactin-producing E. coli found in the pks-island region (pks+E. coli) has been linked to colorectal cancer risk; multiple studies have shown that colorectal cancer patients frequently harbor higher concentrations of pks+E. coli strains in their colonic mucosa than non-cancerous individuals.50,51 In another study, the researchers used an in-vitro infection model to show that the morphology of the primary colon epithelia (PCE) and colorectal carcinoma (HCT116) cell lines changed significantly both before and after they were infected with pks + E. coli. This finding suggests that pks+ E. coli plays a role in the pathogenesis and progression of colorectal cancer. 52
Bacteroides fragilis and Colorectal Cancer
Bacteroides fragilis (B. fragilis) is an anaerobic gram-negative bacterium that is frequently isolated from biological specimens of patients with abdominal abscesses, diarrhea, peritonitis, sepsis, and endogenous purulent infections. 53 B. fragilis is classified as nontoxigenic B. fragilis (NTBF) or enterotoxigenic B. fragilis (ETBF) depending on the release of the primary virulence factor, B. fragilis toxin (BFT). 54 B. fragilis toxin (BFT) is a 21 kDa zinc-dependent metalloprotease toxin that binds colonic epithelial cell (CEC) receptors to cause the intestinal epithelial barrier to become compromised, which in turn causes inflammation. 55 By stimulating the Wnt/B-catenin pathway through its BFT activities, enterotoxigenic B. Fragilis promotes cell proliferation and growth in the pathophysiology of colorectal cancer. 56 The toxin also enhances the release of inflammatory molecules from the epithelial cells, which triggers carcinogenesis by activating the NF-κB signaling pathway. 56 Also, ETBF has been reported to specifically stimulate signal transducer and activator of transcription-3 (STATA3) in the colon, causing Th17 infiltration and advancement of colorectal cancer. 57 Furthermore, the colonic epithelia cells that attach ETBF to them are stimulated to cleave the tumor-suppressor E-cadherin, which increases the permeability of the cells. 55 Additionally, the ability of ETBF to form biofilms shields the bacteria from immunological reactions and antibiotics, enabling it to live longer in the colon and perhaps even accelerating colorectal cancer. 55
Enterococcus faecalis and Colorectal Cancer
Gram-positive Enterococcus faecalis is a facultative anaerobic symbiotic bacterium that is a member of the lactic acid bacteria (LAB). 58 The bacteria are popularly found in the human mouth cavity, gastrointestinal system, and vaginal mucosa and are typically resistant to drastic changes in environmental conditions. Although E. faecalis is occasionally employed as a probiotic supplement, its potential to disrupt the DNA of colonic epithelial cells makes the microbe harmful and detrimental to the development of colorectal cancer. 59
E. faecalis produces superoxide compounds and oxygen free radicals such as hydrogen peroxide and hydroxyl radical that activate macrophages through a series of redox processes resulting in genomic instability, and colonic DNA damage, and subsequently put the host at risk for mutations and cancer.60,61 Furthermore, it has been shown that E. faecalis produces metalloproteases such as Gelatinase (GelE), which has the direct ability to breach compromise the intestinal epithelial barrier, and cause inflammation. 62 Additionally, it can cause breakdown of monolayer integrity and activate the macrophage matrix metalloprotease 9 (MMP-9). 63 This may be the cause of progenitor cells’ morphological metamorphosis into a migratory phenotype, which in turn aids in the epithelial-mesenchymal transition. In addition, Zhang et al showed in colorectal cancer cell lines that, the tumor-stimulating metabolite biliverdin BV, produced by E. faecalis, produced proliferative and angiogenic effects on colorectal cancer, mostly through PI3K/AKT/mTOR activation pathway. 64
Streptococcus bovisand Colorectal Cancer
Streptococcus bovis, a gram-positive coccus, is found in the normal gut flora of about 16% of adult humans. 65 S. bovis is classified into 2 biotypes, I and II. Biotype I can be distinguished from biotype II by its capacity to ferment mannitol. Compared to other S. bovis subtypes, Streptococcus gallolyticus (S. bovis biotype I) was observed in 71% of colorectal cancer cases, and 49% against 8% of healthy tissues. 66
In vitro studies demonstrate that S. bovis has a cell wall antigen that binds to the colon mucosa’s collagen IV and stimulates the release of pro-inflammatory cytokines including interleukin-1 (IL-1) and interleukin-8 (IL-8). 67 Moreover, it was demonstrated that S. bovis proteins were connected to an in vitro overexpression of cyclooxygenase-2 (COX-2). COX-2 has been shown to overexpress in human colorectal cancers and to promote angiogenesis while inhibiting apoptosis. 68
The Impact of Microbiota-Derived Toxins on Colorectal Cancer Pathophysiology.
DNA Methylation and Colorectal Cancer
The most well-researched epigenetic mechanism, DNA methylation, involves a chemical change in the fifth carbon of cytosine in CpG dinucleotides, which is catalyzed by DNA methyltransferases (DNMTs) and S-adenosylmethionine (SAM) as a cofactor. 72 DNA methylation is regulated by 3 different DNMTs: DNMT1 preserves the methylated state immediately following DNA strand replication, while DNMT3A and DNMT3B encourage de novo methylation, primarily during embryogenesis. 73 In the one-carbon metabolism, the 5-methyltetrahydrofolate metabolite is produced and serves as a methyl donor for the conversion of homocysteine to methionine, which is then metabolized to SAM. This process produces the methyl donor SAM, which is necessary for methylation processes. 74 Moreover, SAM is produced by the metabolism of folate.
The common microbiota that has been researched for its potential involvement in DNA methylation is Fusobacterium nucleatum (F. nucleatum) which has primarily been studied in CIMP-positive tumors. However, certain bacteria species including Hungatella hathewayi (H. hathewavi), Streptococcus spp, Parvimonas micra (P. micra), and Bilophila wadsworthia have also been implicated in DNA methylation. CpG island methylator phenotype (CIMP) is associated with CpG island hypermethylation, which is predominantly present in suppressor gene promoters. High concentrations of F. nucleatum were linked to CIMP positive in a study by Tahara et al, and the phenotype was examined in colorectal cancer tissues using 7 gene markers for CIMP diagnosis; ER, SFRP1, MYOD1, MGMT, SLC16A2, SPOCK2, and N33. 75 Additionally, F. nucleatum is reported to cause hypermethylation in the promoter regions of several tumor suppressor genes such as MTSS1, PKD1, PTPRT, MLH1, CDKN2A, and EYA4 in CIMP+ tumors of colorectal cancer resulting in silencing of these genes. 76 The processes by which F. nucleatum performs its function of methylating DNA are associated with the release of hydrogen sulfide, inflammatory cytokines, reactive oxygen species (ROS), and DNMTs recruitment. 77 Hydrogen sulfide, which is released by F. nucleatum, attaches to E-cadherin and initiates an immunological signaling cascade that results in persistent inflammation and the subsequent release of inflammatory cytokines. The presence of F. nucleatum in immune cells has been linked to aberrant methylation. 77 During an inflammatory reaction, reactive oxygen species generated damaged DNA, particularly guanine, a nucleotide that is prevalent in CpG islands. The damaged guanine produces 7,8-dihydro-8-oxo-guanine, which recruits DNMTs to the damaged site leading to hypermethylation and eventually silencing the genes in that region. 77 It has been shown that F. nucleatum can induce nuclear activity and the expression of DMT1 and DMT3A in 2 distinct colorectal cancer cell lines (HT29 and HT116), as well as DNMT3B in a normal cell line (NCM460). 78
The gram-positive, rod-shaped bacterium Hungatella hathewayi has been linked to hypermethylation in the genes SOX11, THBD, SFRP2, GATA5, ESR1, EYA4, CDX2, and APC. It was reported that colorectal cancer cell lines cultured with H. hathewavi expressed more DNMTs. Therefore, it is postulated that the bacteria recruit DNMTs, which subsequently induces DNA methylation. 78 Thus, this may be the mechanism by which this microbe induces methylation.
The Streptococcus spp, particularly Streptococcus bovis and Streptococcus gallolyticus in particular have been linked to hypermethylation of key tumor-suppressor genes, including MLH1 and APC, which are involved in oncogenesis.11,79 The bacterial production of inflammatory cytokines, which trigger immunological response and modify DNA methylation patterns, is the suggested mechanism of action. It has been demonstrated that the oral pathobiont Parvimonas micra, which is linked to colorectal cancer, causes DNA hyper-methylation of important tumor-suppressor genes including SCIN, HACE1, TSPAN13, FBXO32, IGFBP7, SIX1, or CXXC5. 80
P. micra phylotype A significantly increased global DNA methylation of primary cells, and the capacity of bacterium to cause persistent inflammation and dysregulate host immune responses could be the mechanism for DNA hypermethylation. 80 Furthermore, P. micra may indirectly affect DNA methylation patterns via its metabolites such as lipopolysaccharides (LPS) by interfering with host cell signaling pathways that are known to control DNA methylation processes, such as the TGF-β signaling system and the WNT/β-catenin pathway. 81
The bacterium Bilophila wadsworthia is another species that induces hypermethylation in genes. Studies on the gut microbiota of patients with colorectal cancer have revealed the presence of the Gram-negative, obligately anaerobic bacterium B. wadsworthia and its possible involvement in the development of colorectal carcinogenesis.11,82 According to one study on B. wadsworthia, patients with colorectal cancer who had poorly differentiated tissues had 66% methylation frequency for the secreted frizzled-related protein 2 (SFRP2) gene. 82 Hydrogen sulfide, a genotoxic substance that damages DNA, is produced by the sulfate-reducing bacteria B. wadsworthia. 83 Although no research has connected the hydrogen sulfide produced by B. wadsworthia to DNA hypermethylation, it is assumed that the bacteria use the sulfide it produces to cause DNA hypermethylation.
The Impact of Microbiome on Epigenetic Regulation in Colorectal Cancer.

Gut Microbiota-Derived Metabolites and Colorectal Cancer
Gut bacteria produce several metabolites following the anaerobic fermentation of externally undigested food components. These metabolites interact with the epithelial cells that line the mucosal interface, influencing immune responses and the potential development of numerous illnesses, including colorectal cancer. 84 The metabolites have different properties; some are pro-inflammatory and carcinogenic, while others have anti-tumor and health-promoting properties. The response depends on the regulatory pathways induced during the interaction. Vitamin B and short-chain fatty acids (SCFAs) offer protection against colorectal cancer while other metabolites, such as secondary bile acids (SBAs), have been demonstrated to favor the development of colorectal cancer in the host. 85
Short-Chain Fatty Acids
Short-Chain Fatty Acids (SCFAs) in the colon are derived from bacterial anaerobic fermentation of undigested carbohydrates and consist primarily of acetate, propionate, and butyrate, with a molar ratio of 60:20:20 respectively. 86 A report by Kingkaw and colleagues showed that the predominant bacterial families involved in the production of SCFAs are Bifidobacteriaceae, Enterobacteriaceae, Eubacteriaceae, Lach-nospiraceae, and Ruminococcaceae 87 The most abundant SCFA is the acetate and is produced by several of the gut commensal bacteria including Lactobacillus spp., Bifidobacterium spp., Akkermansia muciniphila, Bacteroides spp., Prevotella spp., Ruminococcus spp., and Streptococcus spp., through the Ljungdahl and the acetyl-CoA pathways. 88 The production of most propionate is by the phylum Bacteroidetes (mostly Bacteroides) and some Firmicutes (Negativicutes) via the succinate pathway, where succinate is converted to succinyl-CoA, and succinyl-CoA is decarboxylated to produce propionate and carbon dioxide using the succinyl-, methylmalonyl-, and propionyl-coenzyme A.89,90 Relatively smaller amounts of propionate are produced through the acrylate pathway; where acrylyl-CoA an intermediate from lactate conversion is reduced to propionyl-CoA by the acrylyl-CoA reducatase and then to propionate89,91 and the propanediol pathway; where 1,2-propanediol is dehydrated to propionaldehyde by glycerol dehydratase and then to propionate by the aldehyde dehydrogenase. 92
Butyrate is the most extensively studied SCFA with the majority of the human gut butyrate-producing bacteria belonging to the phylum Firmicutes, particularly Clostridium, Faecalibacteruim, and Bifidobacterium species. The butyrate produced is usually through the butyryl-CoA:acetate-CoA transferase activity. 93 Some bacteria strains (Intestinimonase) produce butyrate from fermentation of proteins and amino acids via the lysine degradation pathway (Lysine → β-lysine →3,5-Diaminohexanate → 3-keto-5-aminohexanate →3-Aminobutyryl-CoA → butyrate). 94
Gut microbial composition depends on the concentration of SCFAs (usually between 10-130 millimolar), and other environmental factors including diet. 85 Butyrate is the main SCFA rapidly absorbed by the colonocytes, the main source of energy for the colonic epithelial cell, and plays a protective role against colorectal cancer development. 95 Several clinical studies found a decrease in butyrate-producing bacteria species and butyrate in the stool of colorectal cancer patients compared to healthy.96-98
Butyrate has been identified as a ligand for G protein-coupled receptors (GPCRs) including GPR41, GPR43, and GPR109A on colonocytes, and activates downstream signaling pathways. 96 Activation of GPR43 by butyrate inhibits the Wnt/β-catenin pathway that suppresses tumor growth. 99 Interaction of butyrate with GPR109A stimulates the down-regulation of Bcl-2, Bcl-xl, and cyclin D1, and the up-regulation of apoptotic receptor signaling to enhance apoptosis in colonic cancer cells and inhibits signaling of NK-κB.100,101 Reduced concentrations of cytoplasmic glucose-6-phosphate dehydrogenase (G6PD) and membrane glucose transporter 1 (GLUT1) have been shown to occur from butyrate interaction with GPR109A via the AKT signaling pathway. This inhibition of glycolysis and glucose transport ultimately leads to changes in glucose metabolism. The growth and survival of malignant colonocytes can be impacted by changes in glucose metabolism since these cells frequently rely on glycolysis (the Warburg effect), to supply their energy needs and facilitate rapid proliferation. 102 Butyrate has been indicated to inactivate some oncogenic signaling pathways such as mitogen-activated protein kinase 1 (MAPK1) and small mothers against decapentaplegic homolog 3 (SMAD 3) which have been implicated in colorectal cancer development by inhibiting apoptosis and enhancing cancer cells proliferation. 103 As a result of the Warburg effect (where there is an increased uptake of glucose and preferential production of lactate through fermentation even in the presence of oxygen), cancer cells prefer the uptake of glucose instead of butyrate, this results in the accumulation of butyrate by these cancerous cells. Butyrate subsequently enters directly into the nucleus and inhibits histone deacetylase 1 resulting in the reduction of short-chain acyl CoA dehydrogenase (SCAD), which is the main process in mitochondrial butyrate oxidation. 104 Low levels of SCAD reduce mitochondrial auto-oxidation of butyrate and this accounts for the inhibition of tumor growth. Donohoe et al demonstrated that a high level of butyrate is associated with low tumor development in a gnotobiotic mice model treated with butyrate-producing bacteria combined with a high-fiber diet. 105
In a study on colorectal cancer HCT116 cells, the researchers demonstrated that butyrate dose-dependently inhibited the growth of colorectal cancer HCT116 cells by modulating promoter methylation in both the tumor suppressor gene (ABCA1) and oncogene [EGR3]. 106 Butyrate has been reported earlier to upregulate DNA demethylation in the promoter region of DNA mismatch repair (MMRs) genes. 107 Additionally, butyrate triggers apoptosis and reduction in the proliferation of carcinoma cells by inducing the expression of cell-cycle regulators and pro-apoptotic genes via histone acetylation induction as evident in colorectal cancer cell lines.108,109
Vitamin B Complex
The B vitamins including vitamin B2 (riboflavin), vitamin B3 (nicotinic acid), vitamin B6 (pyridoxine), vitamin B9 (folate), and vitamin B12 (cobalamin) known to be produced from food sources, and human microbiota have been associated with low colorectal cancer risk. 110 The B vitamins function as co-enzymes in one-carbon metabolism, important in DNA metabolic activities such as methylation, nucleotide synthesis, and stability and repair. 111 Disturbance in these metabolic activities can lead to abnormalities in DNA methylation, instability in DNA precursors, and deficit in the DNA repair mechanism resulting to carcinogenesis. 111
Vitamin B3 or nicotinamide acid serves as a precursor for the synthesis of coenzymes including nicotinamide adenine dinucleotide (NAD) and nicotinamide adenine dinucleotide phosphate (NADP), essential in several biosynthetic pathways, energy generation and protection from reactive oxygen species. 112 Vitamin B3 has been reported to suppress inflammation and colon carcinogenesis by its interaction with certain GPCRs and prostaglandin receptors.113,114 Vitamin B3 interacts with GPR109A and activates the GPR109A signaling pathway that promotes anti-inflammatory effects of colonic macrophages and dendritic cells, causing them to activate Regulatory T cells (Treg cell). 113 Studies in animal models suggest the effect of vitamin B3 in preventing colon carcinomas, though, the exact molecular mechanism remains unknown. 115
Bile Acids
Bile acids are water-soluble end products of cholesterol metabolism that are produced in the liver and are actively absorbed from the small intestine. In humans, cholic acid (CA) and chenodeoxycholic acid (CDCA) are the 2 main primary bile acids. Bile acids are further metabolized by the intestinal microbiota in the gut into potentially carcinogenic secondary bile acids (SBA) including deoxycholic acid (DCA), lithocholic acid (LCA), and very small amount of Ursodeoxycholic acid [UDCA]. 116 It has been reported that higher concentrations of DCA, LCA, and UDCA were found in the feces of colorectal cancer patients than in healthy individuals, 117 as well as higher concentrations of fecal bile acids. There is evidence that cholic acid and chendodeoxycholic acid can cause colorectal cancer by modifying the signaling pathways of NF-κB, Janus kinase 2 (JAK2), and signal transducer and activator of transcription 3 (STAT3). 118 Studies have also shown that malignant transformation of colorectal adenomas may be mediated by the interaction of bile acids and gut microbiota, with higher levels of secondary bile acids, particularly deoxycholic acid (DCA), being crucial in this process. 119 Deoxycholic acid is one of the main secondary bile acids produced by Clostridium that is known to promote tumor growth in colorectal cancer. 119
Several associated carcinogenic signaling pathways that DCA uses to promote the growth of colorectal cancer have been found. 120 First, in a ligand-dependent manner, deoxycholic acids induced tyrosine phosphorylation and activated the EGFR signaling cascade of tyrosine kinases, which in turn initiated the RAS-extracellular-signal regulated kinase 1/2 (ERK1/2) signaling. To facilitate cell division and proliferation, ERK1/2 may activate activator protein 1 (AP-1). 121 Furthermore, Wnt signaling triggered by deoxycholic acid can trigger an inflammatory response and encourage colorectal cancer cell proliferation, both of which are critical for the development of colorectal cancer. Deoxycholic acid also can impact the growth of cancer by releasing β-catenin, which then enters the cytoplasm, moves to the nucleus, and activates transcription factors including the T cell factor/lymphoid enhancer factor (TCF/LEF) family. 122 Additionally, protein kinase C (PKC) is activated when deoxycholic acid perturbs the plasma membrane and causes NF-κB and p38 mitogen-activated protein kinase (p38 MAPK) to become activated. These 2 proteins are essential for controlling inflammatory and immunological responses. NF-κB that has been activated moves into the nucleus and helps to express IL-1β there. 123
Dietary Mediators of Colorectal Cancer Pathogenesis and Development
Numerous epidemiological studies indicate that diet-related factors are a significant moderator of colorectal cancer.3,124 As a result, it has been noted that the incidence of colorectal cancer is 20%-25% lower in nations with healthy dietary habits. 125 Research has indicated that increase consumption of a “westernized” diet, which is rich in processed and red meat, high in saturated fat and refined sugars, and low in whole grains, fruits, vegetables, whole grains, and fish increases one’s risk of colorectal cancer development. 126 However, a lower risk of colorectal cancer has been linked to following a Mediterranean diet, which is characterized by a high intake of fruits, vegetables, legumes, nuts, grains, fish, and olive oil and a low intake of meat and dairy products. 127
In general, the Westernized diet is a conglomeration of dietary components that can increase the risk of colorectal cancer via multiple pathways, such as oxidative stress, microbial dysbiosis, and inflammation. According to a study by Castello et al, a Mediterranean diet has a preventive effect against colorectal cancer development, while a Westernized diet has a harmful effect. 128
Diet Rich in Red and Processed Meat
In recent decades, varied epidemiological studies have shown the increased intake of red and processed meat, and the associated risk of colorectal cancer development. 129 A recent meta-analysis study has estimated that each 100 g/day rise in consumption of red meat was associated with a 17% higher risk of cancer, 130 and even a greater risk was projected for chemically treated processed meat. 131 There are several theories which explain the negative impact of red meat on cancer development, and this has been attributed to the heme iron reservoir in red meat and the generation of secondary bile acids. Heme iron in red meat acts as a nitrosylation agent forming N-nitroso compounds (NOCs) which form electrophilic species through the lipid peroxidation pathway, and these species eventually methylate the DNA to produce N-methylated purines and O6-methylguanine, with the later having both carcinogenic and mutagenic effect.132,133 Heme iron from red meat also boosts the body’s natural NOC production, particularly N-nitrosodimethylamine (NDMA) and N-nitrosopyrrolidine (NNAL), which have been linked to cause alkylating damage and alterations in genes like KRAS that promote the growth of colorectal cancer.134,135 Additionally, continuous generation of bile acids through repeated consumption of red meat might contribute to the development of colorectal cancer through the generation of reactive oxygen and nitrogen species which inhibits DNA repair pathways and enforces resistance to apoptosis as a result of DNA damage.136,137 The processing of meat at high temperatures for a long time (barbecuing, grilling, and pan-frying) produces compounds such as heterocyclic amines (HCA) and polyaromatic hydrocarbons (PCA) that are potent carcinogens and have been strongly linked to colorectal cancer pathogenesis. 138
Despite the unanimous agreement on the correlation between red meat and the risk of colorectal cancer, no positive correlation has been found with other sources of protein except for red and processed meat. In a study conducted on meat subtypes and their association with colorectal cancer risk, poultry, and pork protein intake showed no association with increased colorectal cancer risk. 139 Substitution of red meat protein with other animal proteins showed a reduced risk of developing colorectal cancer. 140
Fiber-Rich Diet and Colorectal Cancer
Consuming fruits, vegetables, whole grain cereals, beans, and nuts can provide the necessary amount of fiber required by the human body. Fiber has a protective effect against cancer, with studies showing a positive result of up to a 25% decrease in cancer risk for intakes between 33.1 and 12.6 grams per day.141,142 Other studies further showed that intake of fiber 3 times a day can also lead to a 17% decrease in cancer risk particularly for colorectal cancer, and sufficient evidence has underscored the protective role of fiber.143,144 There are several proposed protective mechanisms for fiber on colorectal cancer development include increasing fecal bolus, formation of phytochemicals, bacteria fermentation of fiber to produce short-chain fatty acids (SCFAs), slowing down gut transit time, and delaying absorption of complex sugars. 145 Increased fecal mass and shorter gut transit due to fiber intake allow dilution and time reduction of carcinogenic substances in contact with gut epithelial cells. 146 Additionally, it has been demonstrated that the formation of phytochemicals like lignans, flavonoids, carotenoids, and phenolic acids modulates several major inflammatory signaling pathways, such as NF-κB, MAPKs, STAT, and Nrf-2 signaling, thereby reducing the expression of genes involved in oxidative stress and pro-inflammatory cytokines. 145 The SCFAs produced from fermented fiber in the gut by specific microbiota are known to block the conversion of primary bile acids to secondary acids (deoxycholic acid and lithocholic) which have been implicated in colorectal carcinogenesis. 147 SCFAs are also essential sources of energy for colonocytes and can activate G-protein coupled receptors to regulate cellular differentiation, proliferation, metabolism, signaling cascades, and immune responses. 148 Fibrous fruits and vegetables may also exhibit an extra anti-colorectal cancer effect due to several protective substances they contain such as folic acid, flavonoids, polyphenolic compounds (including epigallocatechin-3-gallate), selenium, calcium, vitamins A, D, E, and C. 149 Folic acid (vitamin B9) has been demonstrated to have a protective effect against colorectal cancer when balanced at physiological levels through diet. 150 In addition, fiber intake changes the composition of gut microbiota by enriching SCFA-producing bacteria including Bifidobacteriaceae, Enterobacteriaceae, Eubacteriaceae, Lach-nospiraceae, and Ruminococcaceae among several others. 87
High Fat Diet
Several preclinical studies conducted both in vitro and in vivo have provided evidence supporting the idea that a diet high in fat (HFD) increases the risk of developing colorectal cancer with the majority of the adult population exceeding the recommended level of dietary saturated fat.151,152 The mechanisms by which HFD increases the risk of developing colorectal cancer are not fully understood, though the risk may be linked to the direct effects of HFD such as inducing inflammation, 153 increasing cellular proliferation, 154 and altering intestinal stem cell function. 155 High-fat diets have been linked to persistent colon inflammation, a known risk factor for the development of colorectal cancer. Colon and rectal inflammation arise from a high-fat diet-induced activation of immune cells and the release of proinflammatory cytokines such as interleukin (IL); IL-1, IL-6, IL-8, and tumor necrosis factor-α (TNF-α) in the circulation and tissues. Furthermore, inflammatory signaling pathways in colonic epithelial cells and immune cells, such as signal transducer and activator of transcription 3 (STAT3) and nuclear factor-kappa B (NF-kB), are triggered by high-fat diets.156,157 These pathways may facilitate the growth of tumor cells in the colon and the synthesis of cytokines that encourage inflammation. 154
A meal high in fat (HFD) can increase the amount of bile acid secreted by the intestines, which is then converted into secondary bile acids by gut microbes such as Clostridium spp. By altering the nuclear bile acid receptor, these secondary bile acids can deepen crypts, promote colon cell growth, and ultimately aid in the development of colorectal cancer. An environment that encourages cancer in the colon and rectum may be produced by changes in the bile acid pool and their subsequent effects on the nuclear bile acid receptor.151,154 A high-fat diet has the potential to cause dysregulation of the Retinol-Binding Protein 4 - Stimulated by Retinoic Acid 6 (RBP4-STRA6) signaling pathway, which is involved in the activation of downstream signaling pathways linked to intestinal stem cell stemness, proliferation, and self-renewal. One such pathways keeps Leucine-rich repeat-containing G-protein-coupled receptor 5 positive (Lgr5+) stem cells intact. The activation of the Lgr5+ pathway leads to the increase in Lgr5+ stem cells and the accumulation of genetic mutations, in turn, promotes the growth and progression of intestinal tumors. In addition, Peroxisome Proliferator-Activated Receptor-delta (PPAR-δ) and Peroxisome Proliferator-Activated Receptor-alpha (PPAR-α) signaling, which is amplified by a high-fat diet, activates fatty acid oxidation (FAO) in intestinal stem cells (ISCs). Colorectal cancers may develop and persist in part due to increased stemness and proliferation of intestinal stem cells.155,158 In addition, HFD may indirectly increase colorectal cancer risk by altering the composition of gut microbiota with a reported shift in the firmicutes to Bacteroidetes, reduction in Lactobacilli spp., and enrichment in Clostridium spp. 159
Global and gene-specific DNA methylation, as well as DNA methyltransferase (DNMT) activity, be altered by high-fat diets, especially those high in polyunsaturated fatty acids (PUFAs) such as omega-6 polyunsaturated fatty acids (PUFAs). 160 Studies have shown the high-fat diet rich in n-6 linoleic acid, an omega-6 fatty acid (n6HFD) suppresses the expression of APC using CpG hypermethylation. This results in an increase in the expression of oncogenic transformation (c-JUN) and overexpression of cyclooxygenase-2 (COX-2) via hypomethylation of prostaglandin-endoperoxide synthase-2 (PTSG-2). These events may result in cell proliferation and an increased risk of colorectal cancer. 161 Studies have also shown that subtypes of fat and fatty acid differentially influence colorectal cancer risk; there is a strong and positive association of high intake of monosaturated fat with colorectal cancer risk; however, cholesterol, vegetable fat or linoleic tend to have no effect.152,162
Dairy Products
Whole milk and other dairy products are rich in calcium and other micronutrients. It has been suggested that dairy products help protect against colorectal cancer due to the high calcium content. 163 A dose-dependent observational study reported that consuming approximately 300 mg of calcium per day from dairy products led to a 9% reduction in colorectal cancer. Calcium works to halt cell proliferation, force differentiation, and apoptosis in gut cells by binding to secondary bile acids and intestinal fatty acids by reducing their tumorigenic exposure to the epithelia cells. 164 In addition to high calcium, dairy products also contain other substances including conjugated linoleic acid (CLA), lactose, butyric acid, sphingolipids, and lactic acid bacteria that have anticancer properties. 165
Conjugated linoleic acids (CLAs) have been demonstrated to significantly reduce colorectal cancer in animal models by inducing apoptosis in cancer cell lines via suppressing PI3K/AKT and ERK signaling pathway as well as suppressing the growth of HT-29 cells by inducing apoptosis as a ligand for the PPAR-c gene in a dose-dependent manner. 166 Lactose in fermented dairy products is also metabolized by lactic acid bacteria in the gut to produce butyrate widely known to have anticancer properties. 167 Fermented dairy products including cheese and yogurt have an inverse effect on colorectal cancer development; it is indicated that there was a 12% reduction in colorectal cancer risk for an increase in consumption of 200g a day of fermented dairy products and an 8% for an increase of 50g a day of yoghurt.168,169 The mechanism of dairy products to reduce colorectal cancer development has been associated with calcium, vitamins, and probiotics found in dairy products. 163 However, the relationship between dairy product intake and colorectal cancer risk becomes complex when it is influenced by genetic variations such as lactase persistence. 170
Effects of Dietary Components on Colorectal Cancer.

Alcohol Consumption and Colorectal Cancer Development
Alcohol consumption has been increasingly recognized as a potential contributor to colorectal cancer pathogenesis within the context of epigenetic regulation. 171 In the study conducted by Tsuruya et al, the researchers showed a potential mechanism involving gut microbes converting ethanol to carcinogenic acetaldehyde (AcH), which causes DNA damage. 172 Importantly, alcoholics exhibit altered gut microbiota, characterized by a depletion of obligate anaerobes such as Bacteroides, Akkermansia, Faecalibacterium, and Roseburia and an increase in opportunistic bacteria such as Streptococcus, Enterococcus, Escherichia, and Lactobacillus.172,173 Furthermore, AcH productivity was observed to be decreased in the feces of alcoholic patients compared to non-alcoholic subjects, suggesting a connection between the gut microbiota alterations observed in alcoholics and their capacity to accumulate AcH from ethanol. 172 These findings highlighted a potential epigenetic pathway through which alcohol consumption might influence colorectal cancer pathogenesis.
Alcohol consumption can induce a spectrum of epigenetic modifications that impact gene expression patterns within colonic and rectal cells. Chronic alcohol consumption was linked to alterations in the one-carbon metabolism pathway, which is pivotal for epigenetic regulation. 174 This alcohol-induced impairment of one-carbon metabolism resulted in significant epigenetic changes associated with cancer development, including abnormal promoter gene hypermethylation, global hypomethylation, and decreased antioxidant defense mechanisms. 174 Ethanol metabolism also generated reactive oxygen species (ROS), leading to alterations in DNA methylation patterns, playing a critical role in cancer development. Additionally, alcohol metabolism products impacted NADH levels, influencing histone modifications, and gene expression changes that further influenced cancer susceptibility. 174 These findings highlight the intricate relationship between alcohol consumption and epigenetic mechanisms, potentially contributing to colorectal cancer development and providing avenues for precision medicine in oncology.
Iron Deficiency Anemia and Colorectal Cancer
The most common hematological symptom in cancer patients is still iron deficiency anemia, which is also one of the leading causes of anemia globally. 175 About 60% of occurrences of colorectal cancer are associated with iron-deficient anemia.176,177 Absolute iron deficiency (AID) and functional iron deficiency (FID) are 2 forms of iron deficiency. Overall, there is a true deficiency of iron, a crucial macronutrient involved in numerous biological processes. Anorexia is brought on by tumors, malnourishment, and occult blood loss, all of which are frequent complications of colorectal cancer, and among the factors that contribute to iron deficiency in the patients. 176 A crucial diagnostic of iron availability is transferrin saturation (TSAT), whereas serum ferritin (SF) provides a measure of iron storage. Iron deficiency occurs when transferrin saturation drops below 20%, which can be further distinguished into absolute iron deficiency (SF <100 ng/mL) and functional iron deficiency (SF >100 ng/mL). 178
Inflammation is typically thought to be the cause of iron deficiency anemia brought on by malignancies like colorectal cancer. It is distinguished by decreased erythropoiesis and iron restriction. 179 Comorbidities, as well as the location and size of the tumor, all play a role in the multifactorial cause of iron deficiency anemia brought on by colorectal cancer. It is well known that iron controls macrophage cytokine production. The mechanism of colorectal cancer caused by iron deficiency anemia involves the production of pro-inflammatory cytokines such as IL-6, IL-1, TNF-α, and IFN-γ, which suppress erythropoiesis and promote iron restriction by increasing the synthesis of hepcidin. 180
Additionally, an imbalance between the pro- and anti-oxidant systems has been linked to iron shortage. 181 Iron deficiency results in decreased levels of both enzymatic and non-enzymatic antioxidant systems, including vitamins A, C, and E, glutathione peroxidase (GSH-Px), catalase (CAT), and superoxide dismutase (SOD). 182 The body’s overall antioxidant capacity decreases as a result of these modifications, which also enhance the production of reactive oxygen species (ROS). DNA damage and mutations are known to be caused by ROS, and this may either directly or indirectly contribute to the development of colorectal cancer. 181
In individuals with colorectal cancer, iron deficiency has also been shown to affect oncological outcomes. In addition to being a poorer predictor of long-term outcome in patients with T3N0M0 stage colon cancer, iron deficiency anemia was found to have a negative impact on tumor staging and disease-free survival in comparison to non-anemic patients. 183 These results imply that iron deficiency anemia may affect the prognosis and results of colorectal cancer. This is most likely due to immune system mechanisms that inhibit tumor growth restriction, poor response to chemotherapy or surgery, and immune system limitation of circulating tumor cells that have the potential to undergo metastasis.
Conclusion and Future Direction
Microbial metabolites and genotoxins produced by the gut microbiota in several molecular pathways have been implicated in colorectal cancer initiation and progression through epigenetic regulation. Gut microbiome dysbiosis characterized by a reduction in beneficial bacteria and an increase in detrimental ones contributes to colorectal cancer through epigenetic modifications underscores the need to address these microbial imbalances. High consumption of red meat especially processed meat and a high-fat diet are associated with colorectal cancer risk. On the other hand, a high diet consumption offers a protective role in colorectal cancer development. Moreover, encouraging higher fiber intake stands out as a practical measure, as it not only safeguards against colorectal cancer development by promoting healthy gut function but also fosters the generation of protective agents like short-chain fatty acids (SCFAs). Alcohol metabolites can lead to dysbiosis in the colorectal microbiome and increased intestinal permeability, resulting in bacterial translocation, inflammation, and immunosuppression.
Several interventional approaches studied have been suggested for modulating gut microbiota such as the use of microbiota-modulating agents including prebiotics, probiotic, antibiotics, and dietary modifications.184,185 However, there are still questions to be answered about the suitability of delivery, the kinetics of efficacy, and longevity of modulations of the gut microbiome with these microbiota-modulating agents and dietary modulations. The gut microbiota of an individual varies geographically and ethically as a result of dietary modulations and lifestyle habits, thus extensive clinical research on larger populations is needed on the microbiome of patients of different geographical locations, ethnicity, race, and sex. With the development of colorectal cancer being more sporadic than genetic, the study of epigenetic changes dependent on microbiome dysbiosis, dietary modulations, and alcohol consumption is needed to provide therapeutic methods for managing colorectal cancer.
This calls for experimental and clinical studies on the use of epigenetic dietary phytochemicals such as curcumin found in turmeric, resveratrol found in red grapes, berries, or peas, and genistein found in soy products to better understand how they positively amplify metabolism among gut microbiome, diet, alcohol, and epigenome. Therefore, continued research in modulating microbiome, diet, and lifestyle changes is imperative to unlock novel therapies that function through epigenetic signaling and translate it into tangible advancements in colorectal cancer care.
Footnotes
Authors’ Contributions
The conception and design: EAT, AB-B, OQ & GK-Z; collection of data: GK-Z, SMA, GA, PAB & KGA; manuscript writing: all authors; editing and final approval of manuscript: all authors.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research received funding from the West Africa Genetic Medicine Center (WAGMC), and the Building a New Generation of Academics in Africa (BaNGA-Africa). The authors solely drafted and finalized the manuscript themselves. All the co-authors have given approval for the submission of this manuscript and are in support of any statement including conflict of interest and funding statement.