
Abstract
DNA mutation is a common event in the human body, but in most situations, it is fixed right away by the DNA damage response program. In case the damage is too severe to repair, the programmed cell death system will be activated to get rid of the cell. However, if the damage affects some critical components of this system, the genetic scars are kept and multiply through mitosis, possibly leading to cancer someday. There are many forms of programmed cell death, but apoptosis and necroptosis represent the default and backup strategy, respectively, in the maintenance of optimal cell population as well as in cancer prevention. For the same reason, the ideal approach for cancer treatment is to induce apoptosis in the cancer cells because it proceeds 20 times faster than tumor cell proliferation and leaves no mess behind. Induction of necroptosis can be the second choice in case apoptosis becomes hard to achieve, however, necroptosis finishes the job at a cost—inflammation.
Cancer development is often thought to be the result of uncontrollable mitosis; actually, the failure of programmed cell death (PCD) should be blamed more. In our daily life, we come across many environmental challenges (e.g., radiation, pollution) and intracellular perturbations (e.g., oxidative stress) that are harmful to our DNA integrity. For the most of time, we are O.K. because our DNA damage response (DDR) program takes care of these problems. DDR is a hierarchically organized kinase cascade, including sensing, signaling, and responding.
1
Based on the nature and severity of the damage, the cell selects different tools to fix it. For instance, if the damage happens to only one of the DNA strands, the checkpoint kinase ATR (ATM and RAD3-related) will be activated to repair it by base excision, nucleotide excision, or mismatch repair. However, if both strands are damaged simultaneously, two checkpoint kinases, ATM (Ataxia telangiectasia mutated), and DNAPK (DNA-activated protein kinase) will deal with the problem through either homologous recombination or non-homologous end-joining. Most damages can be fixed in such a way immediately, but some are too severe to repair using any of these strategies. In this case, a signal will be sent to the PCD system, which will eliminate the cell by apoptosis or necroptosis, or other forms of cell death. On the other hand, if the damage happens to any of the genes responsible for DDR or PCD or both, the situation may not be managed properly. Then, the damaged DNA becomes replicated through mitosis. Over time, more and more such damages are accumulated in the genome, resulting in a cancer once they break the threshold (Figure 1). Cancer derives from the failure of DDR or RCD or both. Many extracellular (e.g., radiation), as well as intracellular (e.g., ROS) factors, can cause DNA damages, but most of them are taken care of instantly by the DNA damage response program. If the damage is beyond fixation, programmed cell death will be initiated through the formation of PIDDosome, DISC, apoptosome, or necrosome. As a result, the cell dies of apoptosis mediated by CASP2, CASP8/10, or CASP9. Alternatively, the cell could die of necroptosis if apoptosis fails. However, if the DNA damage response system and the programmed cell death system both fail, the cell containing mutated DNA multiplies through mitosis and is likely to become cancer someday.
This review covers the general concept of cell death and its classification, particularly apoptosis and necroptosis as well as their association with cancer and cancer treatment. Despite the huge collection of publications on these two forms of cell death ( > 800 thousand up to date), some newer investigators still do not have a clear understanding of them. This is reflected by the articles that we have reviewed over the years as well as some published articles in the literature. This current review targets this population of authors and some beginners who are interested in cell death. For this purpose, we have tried to use common language as much as possible throughout the paper so that even an outsider can benefit from reading it.
Death Makes Room for Life
“The goal of all life is death.” - Sigmund Freud
As multicellular organisms, we rely on individual cells in our body to survive, while each cell at any moment of its life has three choices to make: to proliferate, to differentiate, or to die. The earliest documentation about cell death can be traced back to 1842 when German scientist Vogt examined the embryogenesis of a toad and noticed that some cells were “resorbed” and “replaced.” 2 Although some cells do die purely by accident (e.g., tissue injury), the majority of cell deaths take place in a regulated manner. From a fertilized egg to a ready-to-born multicellular embryo, it is not done only by cell division and differentiation; cell death plays a vital part in this process and continues to be such throughout the entire postnatal life. An average adult human body consists of ∼3.72 x 1013 cells and every day over 50 billion of them die and are replaced by new cells instantly. Without such a magnitude of cell death, as Gerry Melino (the head of the Apoptosis and Cancer Laboratory, University of Leicester, UK) estimated, 3 an 80 year-old person would be having a 16 km long intestine and two tons of bone marrow and lymph nodes. It is cell death that keeps our body running more properly and efficiently.
A Cell can die in Many Ways
“We are all born in the same way but we all die in different ways.” - James Joyce
Regulated cell death can take place in many different ways. If you look up the dictionary, the most popular terms that have been widely used to categorize cell death are these three Greek words:
As our knowledge about cell death grows day by day, we started to realize that using only these three words to cover the entire topic of cell death was simply inadequate. A committee, Nomenclature Committee on Cell Death (NCCD), was established to resolve this issue. Based on the morphological features as well as the molecular characteristics associated with the cell death, the NCCD has had four meetings so far to keep the classification updated. Eight names came on the table at first 4 : apoptosis, necrosis, autophagy, cornification, mitotic catastrophe, anoikis, excitotoxicity, and Wallerian degeneration. However, the last four were suspended at the second meeting for the lack of stand-alone criteria. 5
As more research data became available, the terms of anoikis and mitotic catastrophe came back in discussion during the third meeting.
6
Besides these two, four other phrases were coined to split the remainders of apoptotic cell death: caspase-dependent intrinsic apoptosis, caspase-independent intrinsic apoptosis, extrinsic apoptosis by death receptors, and extrinsic apoptosis by dependence receptors. Meanwhile, the name of necrosis was modified into
Among all,
This meeting also introduced four new names: entosis, natosis, parthanatos, and pyroptosis, to cover some rare cases of cell death.
The most recent NCCD meeting was held three years ago,
22
at which the apoptotic cell death was regrouped under the names of intrinsic (mitochondria-based) and extrinsic (receptor-based), as we have been calling them from the very beginning, while necrosis had a new category called “
Like pyroptosis,
Finally, the name
The latest classification of cell death also kept some old names like pyroptosis, parthanatos, entosis, natosis, autophagy, and mitotic catastrophe.
Apoptosis: Slipping Away Quietly
“Life is to make a big scene and then slip away quietly.” - Jin Yong
Apoptosis is a type of rather a peaceful cell death, comparatively speaking. From the initiation till its finish, dozens of molecules are involved orderly, making a big scene. In the end, however, the corpse of the dead cell quietly splits into pieces, which are neatly wrapped up into a pile of “small bags” (apoptotic bodies), waiting for the phagocytes to come to pick up. Once swallowed by the phagocytes, the materials within these “small bags” are decomposed for recycling. 29 The entire process takes place in a confined apartment (the cell), leaving no mess behind. This disposing process is called efferocytosis, meaning “carrying to the grave” in Latin. Despite all sorts of classifications discussed above, apoptosis, in general, occurs via two pathways, the intrinsic (mitochondria-mediated) and the extrinsic (death receptor-mediated). Sometimes one acts alone, while other times, one leads to the other. Regardless of which pathways to take, it all leads to caspase activation.
Human cells express four classes of caspases30,31: apoptotic initiators (CASP2, CASP8, CASP9, and CASP10), apoptotic executioners (CASP3, CASP6, and CASP7), inflammation initiators (CASP1, CASP4, CASP5, CASP11, and CASP12), and the keratinization regulators (CASP14). We have discussed the last two groups already, which are responsible for pyroptosis and cornification, respectively. The rest of the caspases are all apoptosis mediators. While the initiators are mainly responsible for waking up the executioners, plus a few more substrates perhaps, the executioners take down the cell eventually by cleaving thousands of cellular proteins at specific sites, which either disable them directly (e.g., PARP1 cleavage stopping DNA repair) or enabling others indirectly (e.g., DFFA cleavage activating DFFB). Among the four apoptotic initiators, CASP2 and CASP9 mediate intrinsic apoptosis, whereas CASP8 and CASP10 initiate extrinsic apoptosis (Figure 1). From gene knockout studies, we have learned that not only CASP3, 32 but also CASP8 33 and CASP9 34 are all critical for embryonic development, indicating that both intrinsic and extrinsic apoptosis are essential for life.
Intrinsic apoptosis aims to activate the CASP9-initiated caspase cascade (Figure 2). CASP9 activation requires to build an apoptosome in the cytoplasm using cytochrome c and APAF1 (apoptotic protease-activating factor 1) as the basic materials, however, ∼80% cytochrome c stores in the intermembrane space of the mitochondria where it participates in the electron transport between the complexes III and IV of the respiratory chain, and the rest are bound to the inner mitochondrial membrane. The mitochondria outer membrane (MOM) is permeable only to molecules < 5kD, but cytochrome c is an 11.7kD protein. Therefore, for cytochrome c to be in the cytoplasm, bigger holes must be opened in the MOM. This can be done by altering the balance between pro- and anti-apoptotic BCL-2 family members. A lot of cell stress signals, such as radiation, hypoxia, growth factor deprivation, or accumulation of reactive oxygen species (ROS) can trigger this change.
35
In the cytosol, once cytochrome c binds APAF1 to cause dATP hydrolysis to dADP, the subsequent replacement of dADP with the exogenous dATP brings seven APAF1-dATP-cytochrome c complexes together forming an apoptosome.
36
As a result, the CARD (caspase recruitment domain) in APAF1 is exposed to attract CASP9. Once in the apoptosome, CASP9 gets activated through autocleavage. Mitochondrial integrity is critical to the life or death of a cell. Cellular stresses activate p53 to transcribe BAX. The rise of BAX breaks down the mitochondrial outer membrane, letting cytochrome c out to the cytoplasm where it is joined by APAF1 and CASP9 to form the apoptosome. Consequently, CASP9 gets activated to trigger a caspase cascade, resulting in thousands of cellular proteins being degraded. Then, the cell dies of apoptosis. MOMP can be detected using a cationic dye in living cells. In healthy cells, the dye accumulates and aggregates in the mitochondria, giving off a bright red fluorescence. While in the apoptotic cells, the dye cannot aggregate in the mitochondria due to the altered mitochondrial transmembrane potential, and thus it remains in the cytoplasm in its monomeric form, fluorescing green.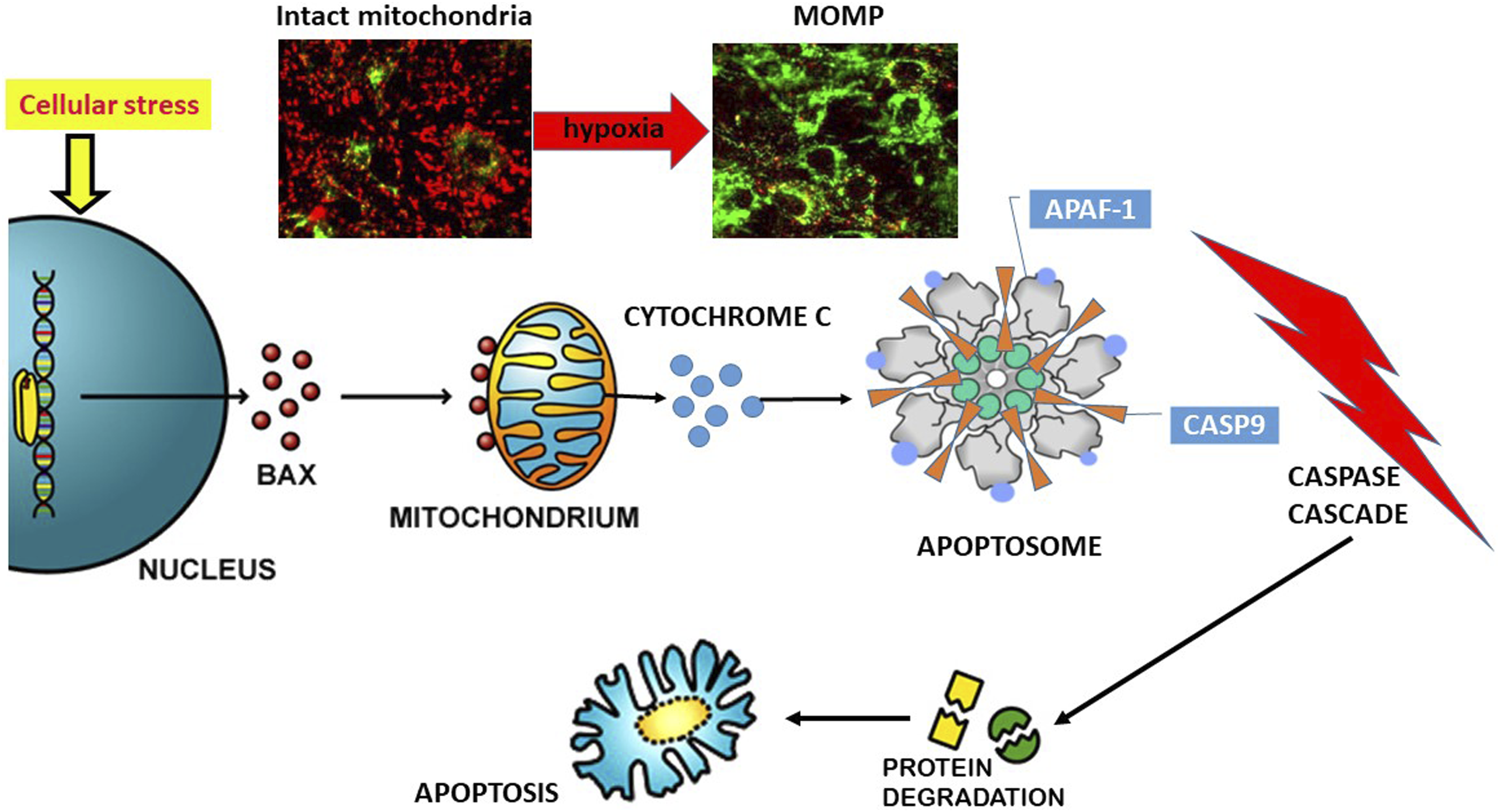
The classification, localization, and partnership of BCL-2 family members.
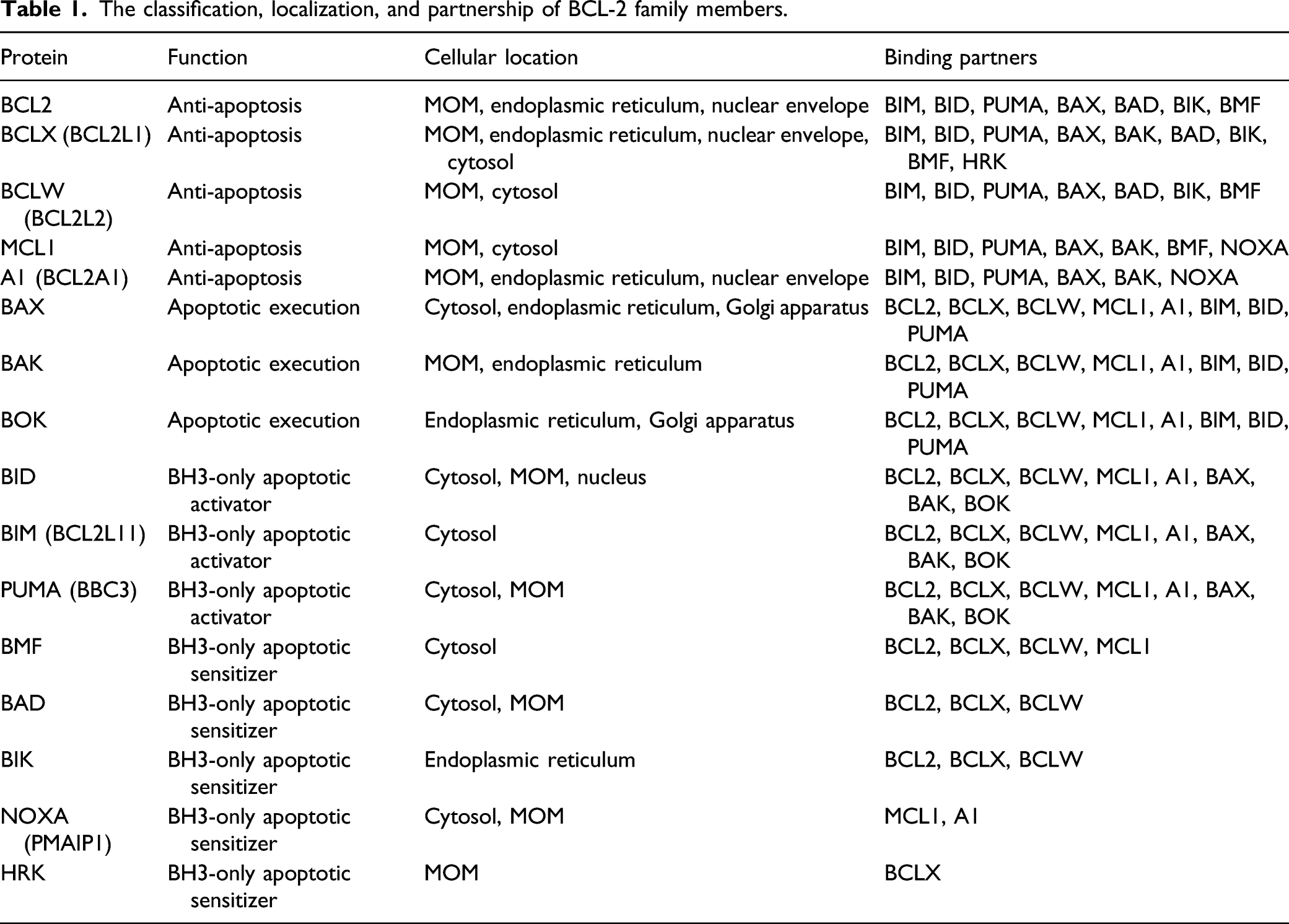
As an apoptotic executioner, BAX (BCL-2-associated X protein) after synthesis is mostly kept in the cytosol in the monomeric form but is constantly trying to land on the mitochondrial wall. When they do, these monomers gather in groups (oligomerization) to open numerous holes in the MOM, letting cytochrome c out to the cytoplasm (Figure 2). BAK (BCL-2 antagonist or killer), on the other hand, hides among the anti-apoptotic BCL-2 members on the MOM and is ready to split the wall open at any moment if the “guards” are off-duty. Normally, the anti-apoptotic BCL-2 proteins keep the “attackers” away from the powerhouse by forming transient heterodimers with BAX/BAK. 41 BOK (BCL–2-related ovarian killer) is the least known member in this category. Despite its 80% homology with BAX and BAK, BOK is mostly found on the endoplasmic reticulum. 42
Upon an apoptotic signal, some BH3-only proteins are activated first to transduce the signal to the apoptotic executioners either through direct binding or by taking away the anti-apoptotic partners from them indirectly. 43 Among all the BH3-only proteins, BID (BH3 interacting domain death agonist), BIM (Bcl-2-interacting mediator of cell death), and PUMA (p53-upregulated modulator of apoptosis) are known as the “apoptotic activators,” which are capable to bind not only all of the anti-apoptotic BCL-2 members to inactivate them but also the apoptotic executioners to activate them, while the rest of the BH3-only proteins, or “apoptotic sensitizers,” have to take an alternative route to reach this goal, namely, by liberating the “activators” from the possession of the anti-apoptotic Bcl-2 members. The “apoptotic sensitizers” are also diversified in their capability. For instance, BMF can bind to all of the anti-apoptotic BCL-2 proteins except A1, BAD and BIK bind three of them (BCL2, BCLX, and BCLW), NOXA favors two (MCL1 and A1), while HRK is only interested in BCLX. Each “activator” has its preference for the apoptotic executioners. For instance, BID is required for BAK activation while BIM supports BAX. Nevertheless, recent studies showed that except for BAD, all of the BH3-only proteins can function as activators sometimes.44,45 Once the anti-apoptotic BCL-2 members are captured by the BH3-only proteins, it takes only ∼5 min for BAX/BAK to permeabilize the entire mitochondrial population of the cell (Figure 2).
In addition to cytochrome c, the mitochondrial outer membrane permeabilization (MOMP) also releases several other molecules that are stored within the intermembrane space, including SMAC/DIABLO, HTRA2, and AIF. 46 While cytosolic AIF induces parthanatos, SMAC/DIABLO (second mitochondria-derived activator of caspase) and HTRA2 (HtrA serine peptidase 2) bind to IAP family members (XIAP, survivin, etc.) to remove their inhibitory effect on caspases. 6 The activated CASP9 cleaves and activates CASP3/7, which in turn cleaves thousands of cellular proteins, resulting in cell death. Therefore, MOMP represents a “point-no-return” in the life of a cell. No cell can stay alive when its entire mitochondrial population is permeabilized.
In addition to CASP9-mediated classic intrinsic apoptosis, CASP2 by itself can also induce intrinsic apoptosis. CASP2 is an intermediate caspase between the initiators and the executioners.47,48 Structurally, it has a CARD-containing prodomain-like CASP9, while functionally it cleaves BID like CASP8/10 on one hand and cleaves PARP1 and many other proteins like CASP3/7 on the other hand. 49 Like CASP9 and CASP8/10, which are activated by the formation of apoptosome and death-inducing signaling complex (DISC), respectively, CASP2 is activated via the formation of PIDDsome, 50 a complex consisting of PIDD1 (p53-induced protein with death domain) and CRADD (CASP2-and-RIPK1 domain-containing adapter with death domain). PIDD1 is a 100kD protein that is constitutively degraded into three functional fragments: PIDD-N (48kD), PIDD-C (51kD), and PIDD-CC (37kD). In response to mild DNA damage, PIDD-C translocates to the nucleus where it forms an association with RIPK1 (receptor-interacting protein kinase 1) and NEMO (inhibitor of nuclear factor κB kinase regulatory subunit γ). Through subsequent sumoylation, phosphorylation, and ubiquitylation, NEMO brings the complex to the cytoplasm where it degrades IκBα, leading to NFκB activation and cell survival. Upon severe DNA damage, however, PIDD-CC becomes the predominant form, which binds to CRADD via their death domains, and then through CRADD binds CASP2 using their common CARDs. In this way, CASP2 becomes activated to initiate apoptosis. 51
In contrast to intrinsic apoptosis, extrinsic apoptosis is mediated by a group of type I transmembrane proteins known as the death receptors, 52 which is a subcategory of the TNFR superfamily. These receptors share a common intracellular sequence named the death domain (DD). Upon ligation by a specific ligand, the death receptor configures its DD to recruit other DD-containing cytoplasmic proteins to build a platform either to fight for cell survival through NFκB or to make a sacrifice through apoptosis.
Death receptors vs decoy receptors.

While DD is essential for extrinsic apoptosis, at least another 30 cytoplasmic proteins have been found to contain similar structures. Ironically, the majority of these cytoplasmic proteins have little to do with cell death. 53 NFκB/p100 and NFκB/p105 are two good examples. As we know, they promote cell survival rather than death. Nevertheless, a few of them are involved in the apoptotic process by serving as adapters between the death receptors and the downstream players. 54 For example, TRADD (TNF receptor-associated death domain) and RIPK1 help TNFR1, DR3, DR6, and NGFR to build the signaling Complex I or Complex II. Complex I supports cell survival via NFκB activation while Complex II can lead to either apoptosis by activating CASP8/10 or necroptosis by activating RIPK3. FADD (FAS associated via death domain), on the other hand, is the only adapter protein that carries the “license to kill.” 55 FADD contains a death effector domain (DED) that fits the same structures in CASP8/10 and thereby activating these initiator caspases directly by forming a DISC, an equivalent structure to apoptosome and PIDDosome that we have discussed above. Among all the death receptors, FAS, DR4, and DR5 use FADD as their primary adapter to signaling. For the other death receptors, FADD has to wait till Complex I dissociating from the cell membrane and turning into Complex II in the cytosol to be a part of the signaling. In another word, the only way for TNFR1, DR3, DR6, or NGFR to induce cell death is when the DD of TRADD or RIPK1 becomes available for FADD to bind. To these four receptors, RIPK1 is like a “lieutenant governor” to TRADD, assisting TRADD to build Complex I and II and taking over the job when TRADD is unavailable. Only when NFκB-supported cell survival and caspase-supported apoptosis both fail, will RIPK1 be activated to form its own “gang” with RIPK3 and MLKL, resulting in necroptosis.
In addition to the death receptors, tumor cells often choose to express some proteins that resemble the death receptors only without DD. The tumor cells use these molecules to keep the cognate ligands away from the real death receptors. We call them the decoy receptors. Four such proteins have been identified, including DCR1 (TRAILR3, TNFRSF10 C), DCR2 (TRAILR4, TNFRSF10D), DCR3 (TNFRSF6B), and OPG (TNFRSF11B). Among them, DCR1, DCR2, and OPG bind to TRAIL (TNFSF10) to block its interaction with DR4 or DR5. OPG can also bind to RANKL (TNFSF11) with a greater affinity to block the RANK (TNFRSF11A) signaling. DCR3 also has multiple partners. 56 It can bind to TL1A (TNFSF15), FASL (TNFSF6), or LIGHT (TNFSF14) to block their interactions with DR3, FAS, HVEM (TNFRSF14), and LTBR (TNFRSF3), respectively. Overexpression of decoy receptors is a common strategy for tumor cells to escape from extrinsic apoptosis.
Necroptosis: Falling Into a “Trap Door” and Landing Explosively
“If plan A fails, remember there are 25 more letters”. - Chris Guillebeau
As a default mechanism to keep the cell population optimal, apoptosis runs through embryonic development as well as the entire postnatal life. It is also the primary mechanism to suppress cancer growth. Apoptosis eliminates the “sick” cells peacefully without disturbing the homeostasis in the neighborhood. As soon as the process is finished, the corpse of the dead cell is immediately decomposed and recycled by the phagocytes through efferocytosis. For this reason, apoptosis is barely detectable in vivo, even though it happens frequently. However, most cancer cells have developed ways to escape from apoptotic cell death. Take the esophageal cancer cells as an example. They often overexpress the decoy receptors to compete with the death receptors for the common ligands.57,58 In this way, they lower the chance to become a victim of extrinsic apoptosis. In addition, they rejuvenate themselves by expressing embryonic genes, e.g., survivin, to keep caspases inactive. 59 Even when these barricades are removed using molecular techniques (e.g., gene manipulation or exogenous stimulation), these cancer cells can still escape from death by overexpressing TRADD. 60 As we know, TRADD preferably mediates NFκB activation and cell survival rather than cell death. Overexpression of TRADD can lower the possibility of FADD-mediated cell death. Furthermore, > 50% of tumor cells are found to carry TP53 mutation. As we know, p53 controls at least 10% of our entire genome, including both pro-survival and pro-death genes, and overlooks any abnormal activities taking place in the cellular world. When a normal cell is about to turn into a cancer cell, p53 is the first to know. 61 Most TP53 mutations generate mutants that only support cell survival, losing its role as a gatekeeper. In this way, the cancer cells would be able to propagate without being detected. Fortunately, the human body has set up a “trap door” for the “escapers”, in case apoptosis fails to stop them. That is necroptosis (Figure 1), our second line of defense against malignancy.62,63
The best-studied case of necroptosis is TNFR1-mediated cell death when NFκB and CASP8 both are dysfunctional. TNFR1 is a 55kD transmembrane protein expressed ubiquitously. Normally, it is kept silent by BAG4 (SODD). Upon TNF stimulation, BAG4 comes off the receptor to expose the DD structure for TRADD to grab.64,65 TRADD then recruits RIPK1 and TRAF2/5 to build Complex I. TRAF2/5 then recruits cIAP1/2 (E3 ubiquitin ligases) to add K63 poly-ubiquitin chains to RIPK1, making it possible to recruit three important complexes to join the team, namely, LUBAC (the linear ubiquitin chain assembly complex), TAK1 (TGFβ-activating kinase 1 complex), and IKK (IκB kinase complex). LUBAC adds M1 linear ubiquitin chains to RIPK1 to further stabilize the platform, while TAK1 phosphorylates IKKβ of the IKK complex, which in turn phosphorylates IκBα, leading to its degradation. Without the inhibition from IκBα, NFκB dimers (p50/p65) are liberated to translocate to the nucleus to transcribe pro-survival genes, such as cIAP1/2, c-FLIP, BCL-2, and BCL-xL. Although RIPK1 seems to be the center of Complex I, it only serves as a building block for the platform and does not have enzyme activity at all. 66
Both TNFR1 and NFκB can keep their activity optimal through negative feedback mechanisms. TNFR1 does this by releasing a fraction of the receptor proteins after synthesis to the extracellular space to absorb some of the ligands before they reach the membrane-bound receptor. In addition, the membrane-bound TNFR1 does the same by constitutively shedding off its extracellular domain containing the binding site for the ligand. 67 The activation of NFκB, on the other hand, is often coupled with the upregulation of A20 and cylindromatosis, and both can de-ubiquitinate RIPK1 and convert the membrane-associated Complex I into a cytosolic Complex II, in which FADD, CASP8, and/or c-FLIP come to the “party.” CASP8 gets activated to digest RIPK1, while c-FLIP works as a decoy to prevent this to happen. For some reason, between CASP8 and c-FLIP, the latter is usually more welcome to Complex II. Therefore, the expression level of c-FLIP determines whether CASP8 can be activated or not.68,69 If c-FLIP is not a part of the Complex II, CASP8 will be activated completely through homo-dimerization, leading to RIPK1 degradation and apoptotic cell death. However, if CASP8 in Complex II is completely replaced by c-FLIP, RIPK1 will gain its kinase activity and bind RIPK3 to form necrosome, initiating necroptosis. On the other hand, if c-FLIP companies CASP8 to the “party” (a more common situation), CASP8 will get activated partially, which can still cleave RIPK1 and RIPK3 but not CASP3. In this case, neither apoptosis nor necroptosis will occur and the cell will go on to live. In either case, RIPK1 appears to be the center of decision-making. For this reason, RIPK1 is often mistaken as the key to necroptosis, and as long as necrostatin-1 (a specific inhibitor for RIPK1) is given to the cells, necroptosis would be impossible to happen. Such publications can be easily found in the literature. The fact of the matter is, even when RIPK1 is omitted completely, at least two other proteins, TlCAM1 (toll-like receptor adapter molecule 1) and ZBP1 (Z-DNA binding protein 1), can activate RIPK3 and induce necroptosis. 70 TlCAM1 is the adapter protein for the toll-like receptor TLR3 and TLR4, while ZBP1 detects foreign nucleic acid. That means, necroptosis is not only mediated through death receptors, pathogen infections can trigger it as well. Regardless of how necroptosis is initiated, RIPK3 activation is always required. Therefore, RIPK3 is the real key to necroptosis. This could be the reason why most cancer cell lines do not express RIPK3 because they would fall into the “trap door” and get killed otherwise. On the other hand, RIPK3-deficient mice do not show increased susceptibility to cancer, which may be due to the existence of the functional apoptotic machinery. After all, apoptosis is the first line of defense against cancer. Several pharmacological inhibitors for RIPK3 have been developed, for example, GSK843 and GSK872, which should be more useful in both laboratory and clinical investigations than necrostatin-1.
After activation, RIPK3 phosphorylates MLKL, which translocates to the plasma membrane where it binds phosphatidylinositol phosphates through oligomerization to cause membrane rupture. As a result, the cellular contents will spill out explosively, stimulating an immune response in the region. Therefore, comparing to apoptosis, necroptotic cell death comes with a price, inflammation. As we know, almost all of the cancers start with inflammation.
Using PCD as a Strategy to Treat Cancer
“Death is the cure for all diseases”. - Sir Thomas Browne
In the battle against cancer, we prefer to eradicate the transformed cells via induction of apoptosis. To achieve this goal, first, we have to restore the paralyzed PCD system in the cancer cells, and meantime, perhaps we can try to lower the threshold for apoptosis as well. As we discussed above, cancer cells are a group of mutants that have successfully escaped from PCD surveillance. One of the common strategies that cancer cells use to do so is to change the balance between pro- and anti-apoptotic members of the BCL-2 family. Such examples can be easily found in breast, lung, prostate, colorectal, gastric, renal, hepatocellular, pancreatic, and esophageal cancer.71-74 These cells usually overexpress BCL2, BCLX, or MCL1 to keep BAX/BAK out of the way so that intrinsic apoptosis becomes impossible. Over the years, a great effort has been made to reverse this process in cancer cells. Some effort intended to activate BAX/BAK directly using synthetic compounds, for example, Compound 106 (ZINC 14750348), which has gained promising results at least in animal studies 75 ; while others tried the indirect approaches, that is, using BH-3 mimetics to remove the anti-apoptotic molecules and thereby to liberate BAX/BAK.73,74 These efforts had great successes in the early clinical trials,76-78 but some serious side effects put the drugs on hold. For instance, patients treated with ABT-263 (Navitoclax), a BH-3 mimetic targeting BCL2, BCLX, and BCLW equally, developed thrombocytopenia due to the loss of platelets, 79 as BCLX is essential for platelet generation. To avoid this complication, ABT-263 was later modified into a newer version called ABT-199 (Venetoclax), which blocks BCL2 selectively. 80 This modification has improved the therapeutic efficacy of this approach substantially. On the other hand, cancer cells often overexpress MCL1 to resist chemotherapy. To solve this problem, several drugs targeting this molecule have been developed. 81 However, because MCL1 is required for hematopoietic cells, side effects remain a big challenge.
In addition to the efforts around MOMP induction, using agonist antibodies to activate extrinsic apoptotic pathways has been explored as well. The problem is that activation of death receptors, as we discussed above, does not always lead to cell death. Sometimes, it goes the opposite direction. TNFR1 is a typical example, which leads to NFκB activation instead of apoptosis mostly. 82 Other mediators of extrinsic apoptosis also have their own “minds” when it comes to the issue of cell death. For instance, CASP8 activation is a critical step in the induction of extrinsic apoptosis and thus CASP8 gene deletion has been found in several cancers, including small cell lung carcinoma, medulloblastoma, glioma, gastric, and hepatocellular carcinomas. However, in some other cancers, such as lung, esophageal, colorectal, cervical, and breast cancer,83,84 CASP8 mutations were found to suppress the malignancy. This contradiction could be related to the negative role of CASP8 in necroptosis. As we discussed above, both RIPK1 and RIPK3 are substrates of CASP8. With CASP8 overexpression and activation, necroptosis naturally becomes difficult.
In contrast, studies with TRAIL-mediated apoptosis have generated some better outcomes. 85 TRAIL binds two death receptors (DR4 and DR5) and three decoy receptors (DCR1, DCR2, and OPG). The death receptors are almost exclusively expressed by tumor cells. Theoretically, TRAIL should be an ideal anti-cancer molecule. However, its clinical use did not achieve success as expected. The reason can be several. As demonstrated in our laboratory,57-61 esophageal cancer cells usually express high levels of the decoy receptors to neutralize the ligand before it reaches the death receptors. They also overexpress TRADD to replace FADD in the DISC formation so that even if the death signal passes through the cell membrane it will be converted into a stimulus for survival via NFκB signaling pathway. 60 Therefore, based on our investigation, clinical studies with TRAIL should not only focus on the ligand and receptor but should also pay close attention to the downstream mediators as well.
Closing Remarks
Cell death operated by PCD is critical to both the prenatal and postnatal life of a human individual. It not only helps the body to maintain homeostasis but also removes the risks of cancer. For this reason, PCD is usually found broken in cancer cells. As health care professionals, we set our goal to eliminate cancer cells from our system as much as possible. Current cancer treatment heavily relies on radiotherapy, chemotherapy, and surgical operation, which all cause severe side effects. Apoptosis is a neat way to remove an unwanted cell without disturbing the neighboring tissue. Furthermore, apoptosis finishes 20 times faster than mitosis. It can effectively shut down tumor growth as long as it is precisely targeted. Any component of the apoptotic cascade can be a good spot to hit on. Researchers are welcome to make their choice. If apoptosis is impossible to induce for any reason, necroptosis can be the second choice. Necroptosis is capable to kill cancer cells at a cost, inflammation, because necroptosis results in cell rupture, which releases the intracellular contents to the local area triggering immune responses. Therefore, the strategy of necroptosis induction should be evaluated based on the patient condition case by case. The key is targeting, keeping the downside to minimal and the upside maximal. Abbreviations; AIF—apoptosis inducing factor; APAF1—apoptotic peptidase activating factor 1; ATM—Ataxia telangiectasia mutated; ATR—ATM and RAD3-related; BAK—BCL-2 antagonist or killer; BAX—Bcl-2-associated X protein; BID—BH3 interacting domain death agonist; BIM—Bcl–2-interacting mediator of cell death; BOK—BCL–2-related ovarian killer; CARD—caspase recruitment domain; c-FLIP—cellular FLICE-inhibitory protein; CRADD—CASP2-and-RIPK1 domain-containing adapter with death domain; DAMP—damage-associated molecular patterns; DD—death domain; DDR—DNA damage response; DED—death effector domain; DISC—death-inducing-signaling complex; DNAPK—DNA-activated protein kinase; FADD—FAS-associated protein with death domain; GPX4—glutathione peroxidase 4; IκB—inhibitor of kappa B; IKK—IκB kinase; LUBAC—linear ubiquitin chain assembly complex; MLKL—mixed lineage kinase domain-like protein; MOM—mitochondrial outer membrane; MOMP—mitochondrial outer membrane permeabilization; NCCD—Nomenclature Committee on Cell Death; NEMO—NFκB essential modulator; NFκB—nuclear factor kappa B; PARP1—poly (ADP ribose) polymerase 1; PCD—programmed cell death; PIDD1 - p53-induced protein with death domain; PUMA - p53-upregulated modulator of apoptosis; RIPK1 - receptor-interacting protein kinase 1; ROS – reactive oxygen species; TAK1—TGFβ-activating kinase 1; TNF—tumor necrosis factor; TNFR—tumor necrosis factor receptor; TRADD—TNFR1 associated death domain protein; TRAF—TNF receptor-associated factor
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by Inner Mongolia Research Foundation for Natural Sciences (2021MS08007), Wu Jieping Medical Science Foundation (320.6750.19089-91), and the National Natural Science Foundation of China (81960445).