
Abstract
Autosomal dominant polycystic kidney disease (ADPKD) may be associated with various epithelial malignancies. The most reported ones are papillary renal cell carcinoma (RCC) and clear cell RCC. Only one noninvasive urothelial carcinoma arising in the renal pelvis has been previously reported in the setting of ADPKD in the English literature. A 52-year-old patient with ADPKD and a history of renal transplant presented with a poorly differentiated sarcomatoid neoplasm in his native left polycystic kidney. A recognizable urothelial or renal cell carcinoma differentiation was not identified in the resected neoplasm microscopically. The initial diagnosis for this specimen was challenging on morphology and immunohistochemistry, but targeted next-generation sequencing provided molecular evidence in support of urothelial origin, indicating a hotspot mutation −124 C > T in the TERT promoter (C228 T) and loss of heterozygosity on chromosomes 9p and 8p. This tumor is unique because, to our knowledge, this is the first report of upper tract sarcomatoid urothelial carcinoma in a patient with ADPKD.
Introduction
The relationship between autosomal dominant polycystic kidney disease (ADPKD) and associated cancer is an area of ongoing research. Observational studies such as the one reported by Yu et al have identified nearly double the cancer incidence in 4636 patients (20 years or older) with ADPKD compared to those matched individuals without the disease. 1 The majority of malignancies in patients with ADPKD are clear cell renal cell carcinoma (RCC) and papillary RCC,2,3 but there are also rare reports of renal sarcoma4–6 and sarcomatoid RCC.7–9 Only one noninvasive urothelial carcinoma arising in the renal pelvis has been previously reported in the setting of ADPKD. 3 We report herein on a patient with ADPKD who presented with an unusual and diagnostically challenging sarcomatoid neoplasm demonstrating metastatic spread and aggressive clinical course.
Patient Presentation
A 52-year-old man presented with a 2-month history of increased left flank pain, nausea, vomiting, constipation, gross hematuria, dysuria and 5 kg weight loss. His medical history was significant for ADPKD, with documented liver cysts but without cerebral aneurysms. Four years prior, he progressed to end-stage renal failure and received a cadaveric kidney transplant to the right iliac fossa. The patient had a renal ultrasound 6 months before his admission that showed no solid lesions in his native kidneys, and no subsequent imaging investigations were done. Given his ongoing left-sided flank pain and suspicion of renal infections, the patient had an elective bilateral radical nephrectomy of the native kidneys. Intraoperatively, it was noted that there was significant inflammation around the left native kidney, and large bulky retroperitoneal lymph nodes were found abutting the aorta. The left kidney and the left adrenal gland were removed, and the right adrenal gland was spared.
Gross examination of the enlarged kidneys showed numerous cysts, consistent with ADPKD. The left polycystic kidney measured 27.4 × 19.6 × 15.5 cm and was almost completely replaced by a white-tan, solid mass that measured 17.5 × 15.3 × 15.1 cm (Figure 1A). The right kidney did not show any solid masses (Figure 1B).
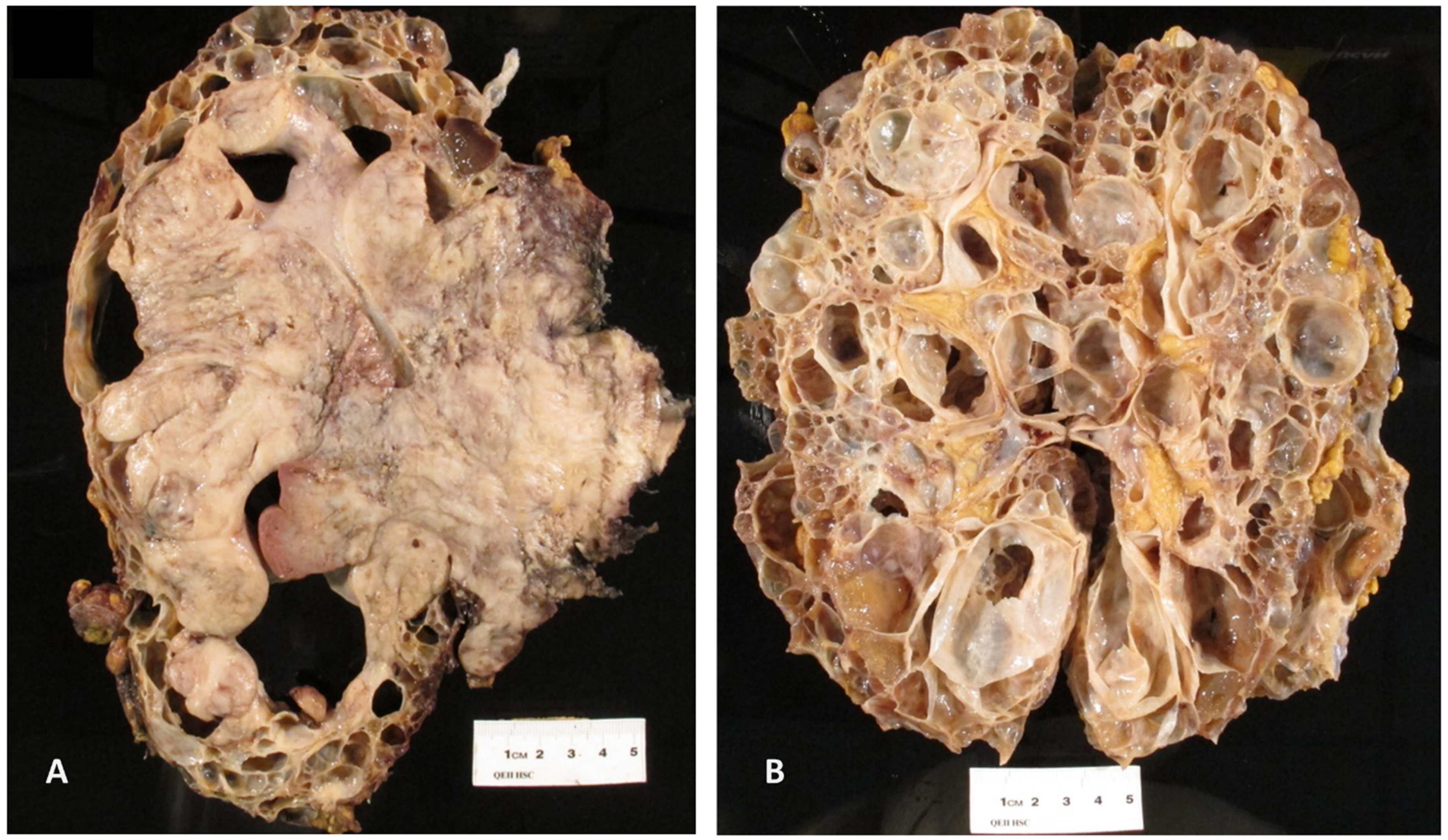
(A) The left polycystic kidney was enlarged and almost completely replaced by a white-tan, solid mass. (B) The right kidney showed typical gross features of ADPKD and did not show any solid masses. Abbreviation: ADPKD, Autosomal dominant polycystic kidney disease.
A subsequent computed tomography (CT) (Figure 2A) scan of the chest showed multiple pulmonary nodules, consistent with metastatic disease, as well as pericardial and pleural effusions and a lytic T11 bony lesion. An enlarged right axillary lymph node was later biopsied, and the biopsy confirmed the presence of metastatic disease. A positron emission tomography (PET) (Figure 2B) scan identified intense uptakes involving multiple organs, including lungs, pleura, lymph nodes, liver, bone, and skeletal muscle. A staging CT of the head showed no evidence of intracranial lesions.
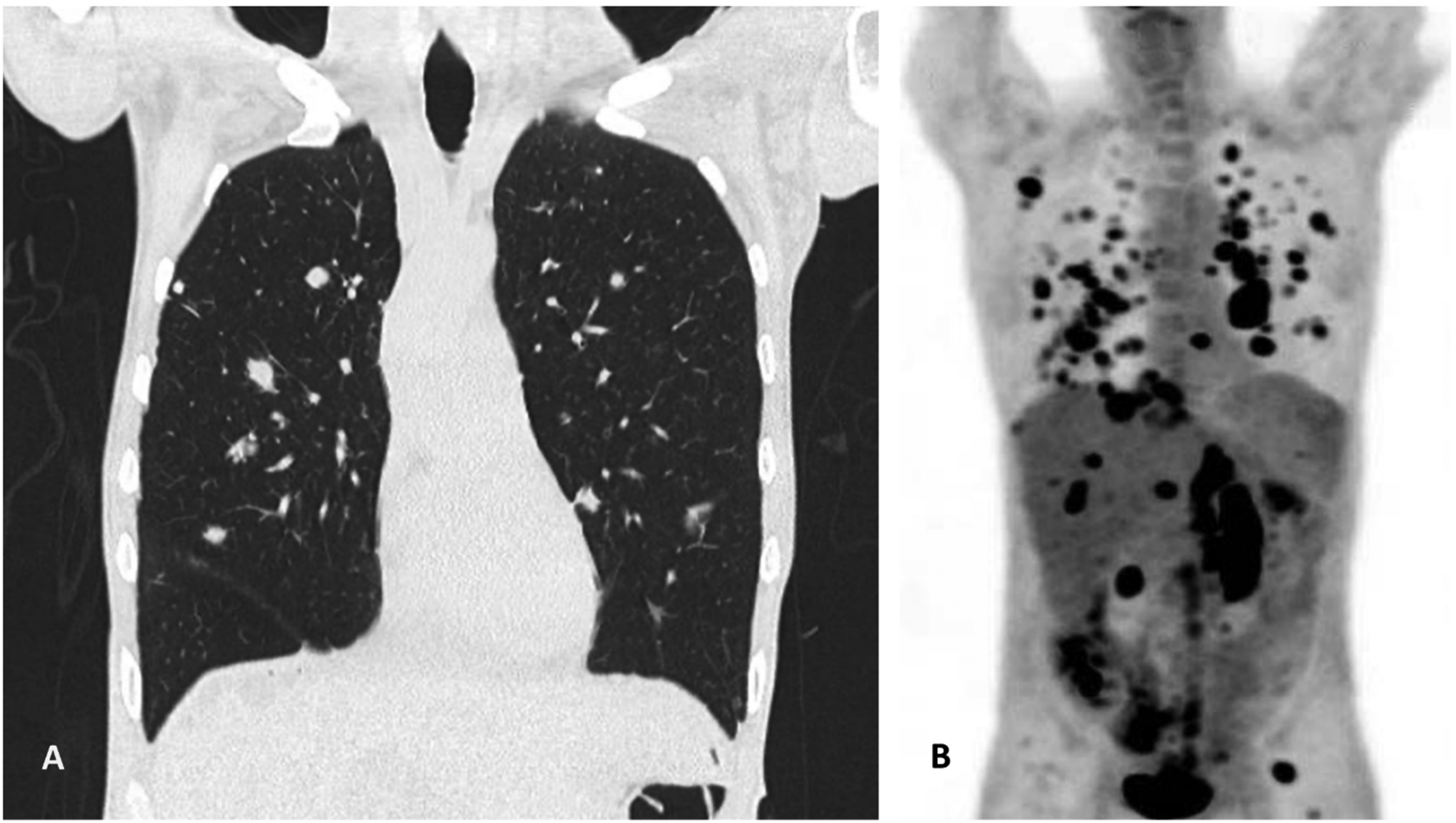
(A) On CT imaging of the chest, there were multiple pulmonary nodules, consistent with metastatic deposits, as well as pericardial and pleural effusions and a lytic T11 bony lesion. (B) PET scan identified intense uptakes involving multiple organs, including lungs, pleura, lymph nodes, liver, bone, and skeletal muscle. Abbreviations: CT, computed tomography; PET, positron emission tomography.
Microscopic examination of the solid mass in the left kidney demonstrated a poorly differentiated neoplasm, mostly composed of spindled cells, with focal areas showing fascicular growth; focal epithelioid and rhabdoid cell features were present with brisk mitotic activity and numerous apoptotic bodies (Figure 3A and B). Focally, there was marked nuclear atypia and pleomorphism (Figure 3C), along with frequent osteoclast-like giant cells that surrounded areas of coagulative necrosis and osteoid formation (Figure 3D and E), in keeping with heterologous differentiation. Importantly, no recognizable components of either urothelial carcinoma or of any RCC type were seen on extensive sampling. The hilar resection margin was involved by neoplastic cells and there was invasion of the renal sinus. The left adrenal gland (Figure 3F) also showed metastatic deposits.
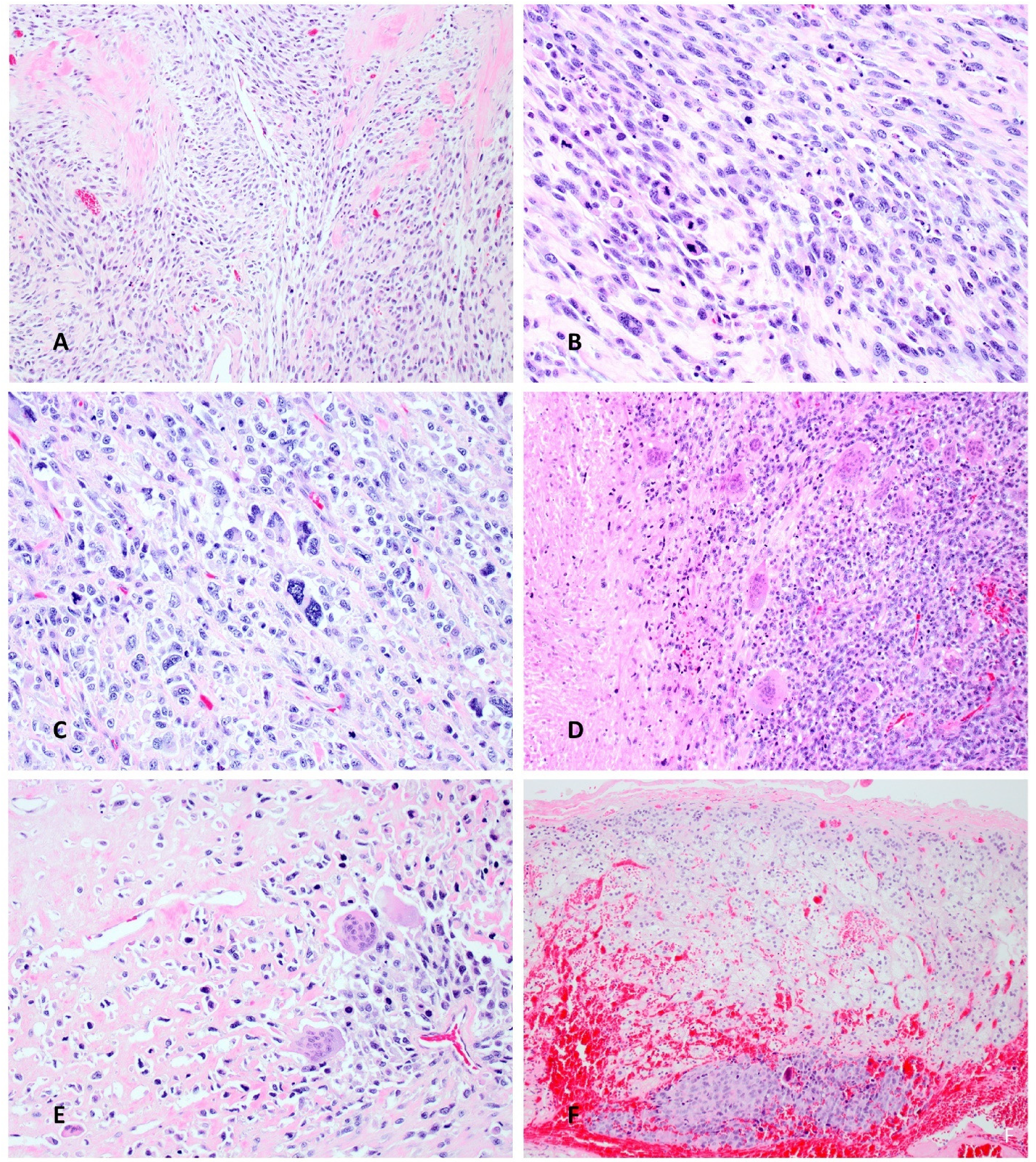
(A) On microscopy, the neoplasm was poorly differentiated and mostly composed of spindle-shaped cells, with focal areas showing fascicular growth. (B) There were brisk mitotic activity and apoptotic bodies, as well as foci with more epithelioid and rhabdoid-like cells. (C) Focally, there was marked nuclear atypia and pleomorphism. (D) and (E) Frequent osteoclast-like giant cells surrounded areas of coagulative necrosis (D, left) and osteoid formation (E), in keeping with heterologous differentiation. (F) Metastatic deposit (lower part of image) was found in the left adrenal gland.
On immunohistochemistry, the neoplastic cells were reactive for vimentin, keratin AE1/AE3 (weak paranuclear), keratin 8/18 (weak paranuclear), GATA3 (focal and weak), INI1, and EMA (focal). The immunostains for keratin 7, keratin 20, keratin 5/6, p63, PAX8, synaptophysin, S100, and MDM2 were all negative. After consultation with external pathology experts, a diagnosis of sarcomatoid carcinoma (or “carcinosarcoma”) of uncertain origin was favored, but the origin of the neoplasm (renal vs urothelial) could not be established. The possibility of retroperitoneal sarcoma, including dedifferentiated liposarcoma, epithelioid sarcoma, synovial sarcoma and leiomyosarcoma, or a malignant peripheral nerve sheath tumor, were also considered in the differential diagnosis.
Next-generation sequencing (NGS) with a comprehensive DNA cancer biomarker panel was performed, targeting the full coding sequence or hotspot regions of 130 genes, 76 microsatellite loci and several single nucleotide polymorphisms (SNP) loci distributed across all chromosomes. NGS revealed a hotspot mutation −124 C > T (C228 T) in the telomerase reverse transcriptase (TERT) promoter and a loss of heterozygosity on chromosomes 2p, 5p, 8p, and 9p (Figure 4). Overall, the molecular findings were supportive of urothelial carcinoma.10–18 Thus, the final diagnosis was sarcomatoid urothelial carcinoma with heterologous osteosarcoma-like differentiation. Owing to advanced metastatic disease, the patient passed away several weeks after the surgical resection.
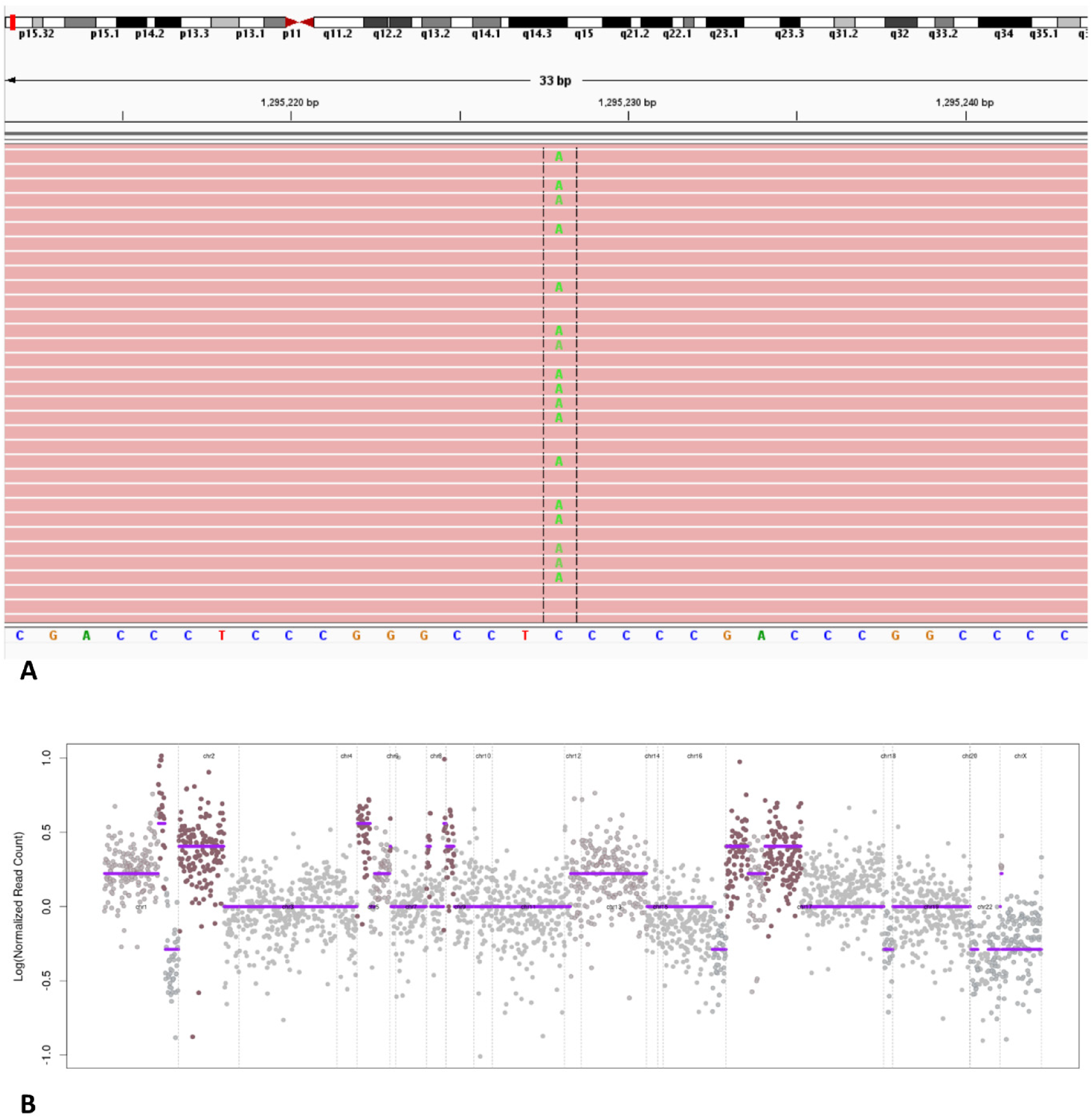
(A) The aligned sequencing reads are visualized in the integrative genomics viewer showing the C > T change in the promoter position chr5:1295228 of the TERT gene. (B) The copy number plot is displayed using the log2 value of the normalized read count for each amplicon included in the NGS panel. This tumor demonstrated markers of aneuploidy with multiple loss of heterozygosity events including the copy number gain of the chromosomal arms 2p, 5p, 8p and 9p. Abbreviation: NGS, next-generation sequencing.
Discussion
Analysis of 301 nephrectomy specimens performed in a setting of ADPKD revealed 16 malignant tumors, including 9 papillary RCCs, 5 clear cell RCCs, 1 multilocular cystic RCC, and 1 noninvasive urothelial carcinoma (pTa) arising in the renal pelvis. 3 Two patients (1 with papillary RCC and 1 with metastatic sarcomatoid clear cell RCC) passed away due to metastatic disease during the follow up. The patient with the upper tract noninvasive urothelial carcinoma was alive during the follow-up period of 19.3 months.
Six previous case reports of renal sarcomatoid tumors arising in patients with ADPKD were also identified in the English literature (summarized in Table 1).4–9 There were 5 male patients and 1 female patient, with ages ranging from 30 to 60 years. All presented acutely with flank pain and/or gross hematuria. All patients were treated with nephrectomy and the final diagnosis was renal sarcoma in 3 patients (1 histiocytic sarcoma, 4 1 rhabdomyosarcoma, 6 and 1 high-grade pleomorphic sarcoma 5 ) and sarcomatoid RCC in 3 patients.7–9 Three of the patients, including 2 with metastatic sarcomatoid RCC7,8 and 1 with histiocytic sarcoma 4 expired during the postoperative period due to complications, failure to thrive, and multiorgan failure, respectively. The patient with rhabdomyosarcoma 6 survived the initial hospital management. The outcomes of the remaining 2 patients were not reported.
Summary of Case Reports of Renal “Sarcomatoid Neoplasms” (Either Sarcomas or Sarcomatoid Carcinomas) Reported in ADPKD.
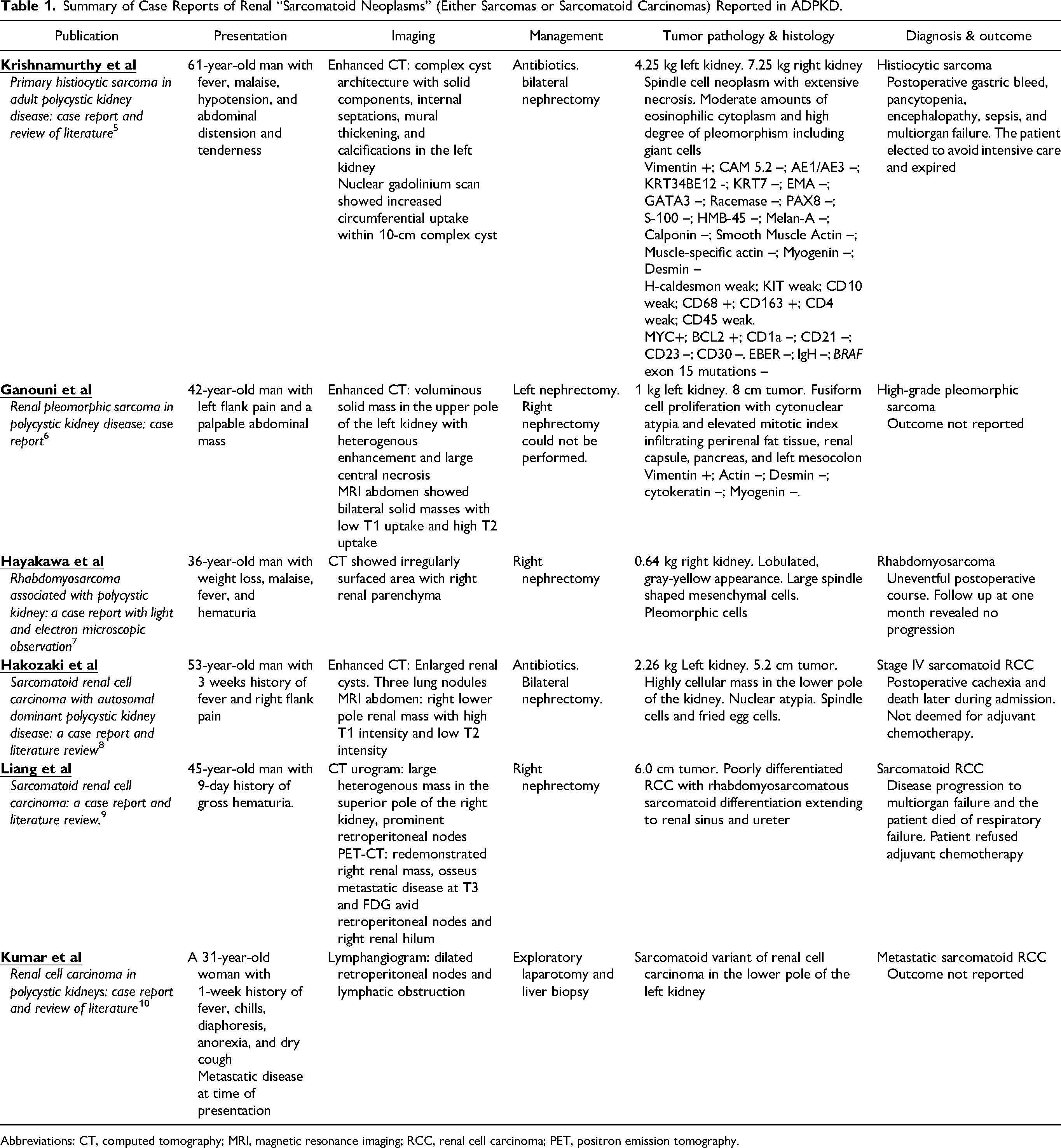
Abbreviations: CT, computed tomography; MRI, magnetic resonance imaging; RCC, renal cell carcinoma; PET, positron emission tomography.
Considering these similar previous reports of sarcomatoid neoplasms arising in patients with ADPKD, the tumor reported herein is unique. First, the tumor was diagnostically challenging and was found in a background lacking detectable kidney parenchyma due to polycystic changes. This made it difficult to ascertain whether the tumor's origin was in the renal pelvis (favoring urothelial carcinoma) or in the renal parenchyma itself (favoring RCC). Secondly, the histology of the tumor was poorly differentiated and lacked a recognizable urothelial carcinoma, urothelial carcinoma in situ (CIS), or RCC component, but showed heterologous osteosarcoma differentiation. Thirdly, the immunostains were not conclusive to differentiate between a sarcomatoid RCC, sarcomatoid urothelial carcinoma or high-grade sarcoma. In tumors like this, the molecular characterization would be the appropriate investigation to potentially resolve such diagnostic issues.
This tumor, to our knowledge, is the first reported ADPKD associated upper tract sarcomatoid urothelial carcinoma with the hotspot mutation −124 C > T in the TERT promoter. TERT promoter region point mutations were recently reported to be associated with bladder cancer.10–12 Wang et al reported a series of 17 patients with upper urinary tract sarcomatoid urothelial carcinoma. 11 Six of the 17 tumors harbored a “hotspot” −124 C > T of the TERT.11,12 Five of the 6 tumors had very minimal (0%) to weak (10%) GATA-3 staining, which is similar to the current tumor. Among the 6 patients with TERT promoter region mutation −124 C > T, all presented with high stage disease: 3 had pT3N2 disease and 3 had pT4 disease. Five of the 6 patients had a very aggressive course and either died after the surgery or were found to have metastasis and/or recurrence of the tumor. These findings may suggest that the TERT promoter region mutation may be associated with advanced cancer and poor prognosis in the upper urinary tract urothelial carcinomas.
Loss of heterozygosity (LOH) of chromosome 9p has also been linked to urothelial carcinoma in multiple reports. Tsai et al investigated 18 high-grade urothelial carcinomas and found 12 of them to have an LOH on chromosome 9p. 13 Ploussard et al detected 9p LOH in 32% of the pT1 grade 3 bladder carcinomas. 14
LOH of chromosome 8p has also been associated with bladder cancer.15,16 It has been linked with urothelial CIS and invasive urothelial carcinoma. LOH of 8p was also found to be associated with urothelial carcinomas of high grade and high stage, as in the current neoplasm.15–18
Summary
We report a sarcomatoid urothelial carcinoma arising in a patient with ADPKD. Most kidney tumors reported in patients with ADPKD have been RCCs, and sarcomatoid malignancies, such as sarcomatoid carcinomas and true sarcomas are exceedingly rare in this setting. Diagnosing this tumor was challenging because the histomorphology and the immunochemistry investigations were inconclusive. This tumor is also unique because, to our knowledge, this is the first reported ADPKD-associated upper tract sarcomatoid urothelial carcinoma with the hotspot mutation −124 C > T in the TERT promoter (C228 T) and LOH on chromosomes 9p and 8p, in keeping with high-grade urothelial carcinoma.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Funding
The authors received no financial support for the research, authorship and/or publication of this article.
Ethical Approval
The Nova Scotia Health Research Ethics Board approves the case report for research use of de-identified information collected from the patient.
Informed Consent
Waiver of consent approved by the Nova Scotia Health Research Ethics Board.