
Abstract
We report an exceptional case of a spindle cell mesenchymal tumor with S100 and CD34 co-reactivity, which harbored a SLMAP::RAF1 fusion. To the best of our knowledge, this is the second case of a spindle cell mesenchymal tumor with S100 and CD34 co-reactivity with this specific fusion. Remarkable is the presence of calcification and heterotopic ossification in the center of our lesion, a feature that, to our knowledge, has not been described yet in RAF1-rearranged spindle cell mesenchymal tumors.
Introduction
Spindle cell neoplasms are difficult to classify, as most exhibit similar morphology without specific immunohistochemical profiles, with a varying risk of malignancy. Molecular genetics have proven a breakthrough in the classification of spindle cell neoplasms as these tumors are associated with kinase-related fusions. 1 NTRK-rearranged spindle cell neoplasms, which fall under the broader term of spindle cell tumors with S100 and CD34 co-reactivity, are a newly defined group within soft tissue tumors that is still emerging.1,2
Here, we describe an adult patient with a spindle cell tumor with S100 and CD34 co-reactivity harboring a SLMAP::RAF1 fusion and showing calcifications and heterotopic osseous metaplasia.
Methods and Results
Clinical Presentation
A 63-year-old woman with a lesion located in the right inguinal canal, detected in 2014 by ultrasound and with echographic follow-up in subsequent years, recently experienced more local discomfort, especially after physical exertion. Magnetic resonance imaging (MRI) of the abdominal wall showed a heterogeneous lesion of 21 × 24 × 27 mm in the inguinal canal encroaching onto the m. abdominus rectus and with extension into the neurovascular bundle (Figure 1). Computed tomographic (CT)-chest/abdomen showed no signs of distant metastases.
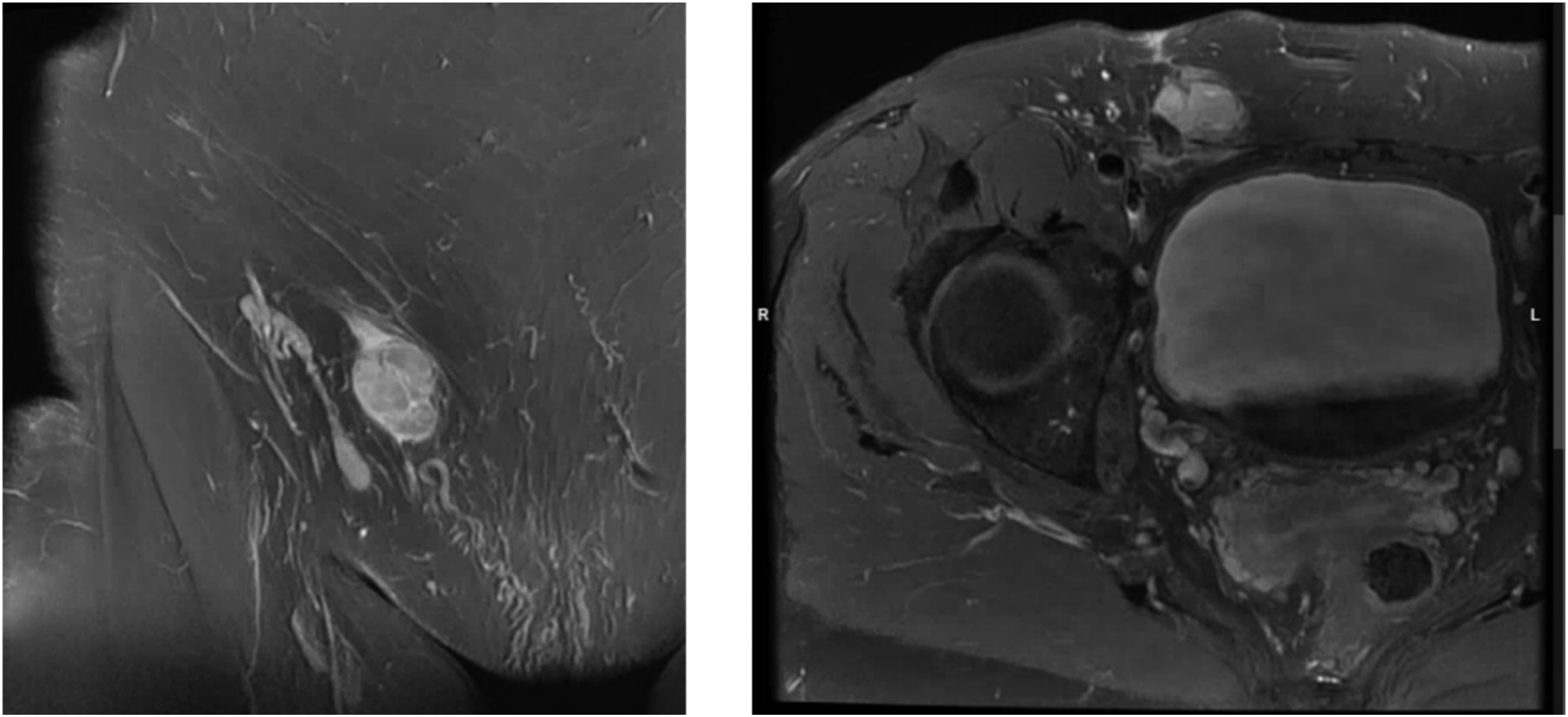
On magnetic resonance imaging (MRI) a lobulated subcutaneous nodularity protruding in the right inguinal canal was seen. There was fairly homogeneous contrast capture of the lesion (Left: coronal MRI T1-weighted image with fat saturation postcontrast; right: axial MRI T1-weighted image with fat saturation postcontrast).
Pathological Features and Immunohistochemistry
A biopsy of the inguinal lesion was performed which showed a mesenchymal proliferation of haphazardly arranged primitive monomorphic spindle cells (Figure 2). The spindle cells had a monomorphic round nucleus demonstrating heterochromatin. There was no high-grade atypia, no brisk mitotic activity, nor necrosis. The stroma was densely fibrous to collagenous. In the background there were hyalinized blood vessels and infiltration by lymphocytes was noted.
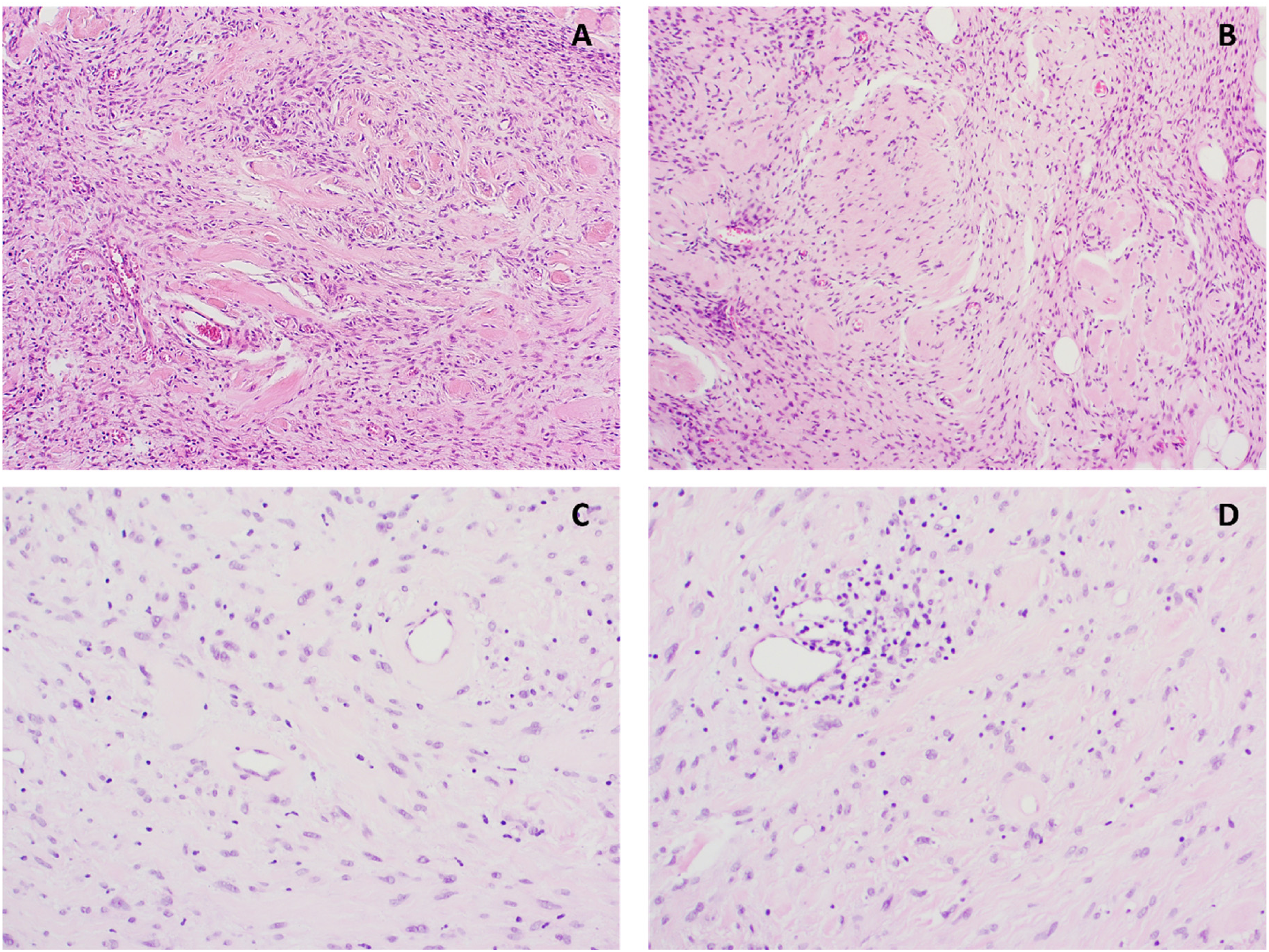
Mesenchymal proliferation of haphazardly arranged primitive monomorphic spindle cells (A, B). The spindle cells had a monomorphic round nucleus. The stroma was densely fibrous to collagenous. In the background there were hyalinized blood vessels (C) and infiltration by lymphocytes were noted (D). (Hematoxylin and eosin [HE], original magnifications 200×).
On immunohistochemistry (Figure 3), the spindle cells were positive for S100 and CD34. There was no expression of SOX10, STAT6, pan-TRK, and ERG in the spindle cells. Beta-catenin staining was cytoplasmic. Expression of H3K27me3 and H3K27me2 was retained in the tumor cells.
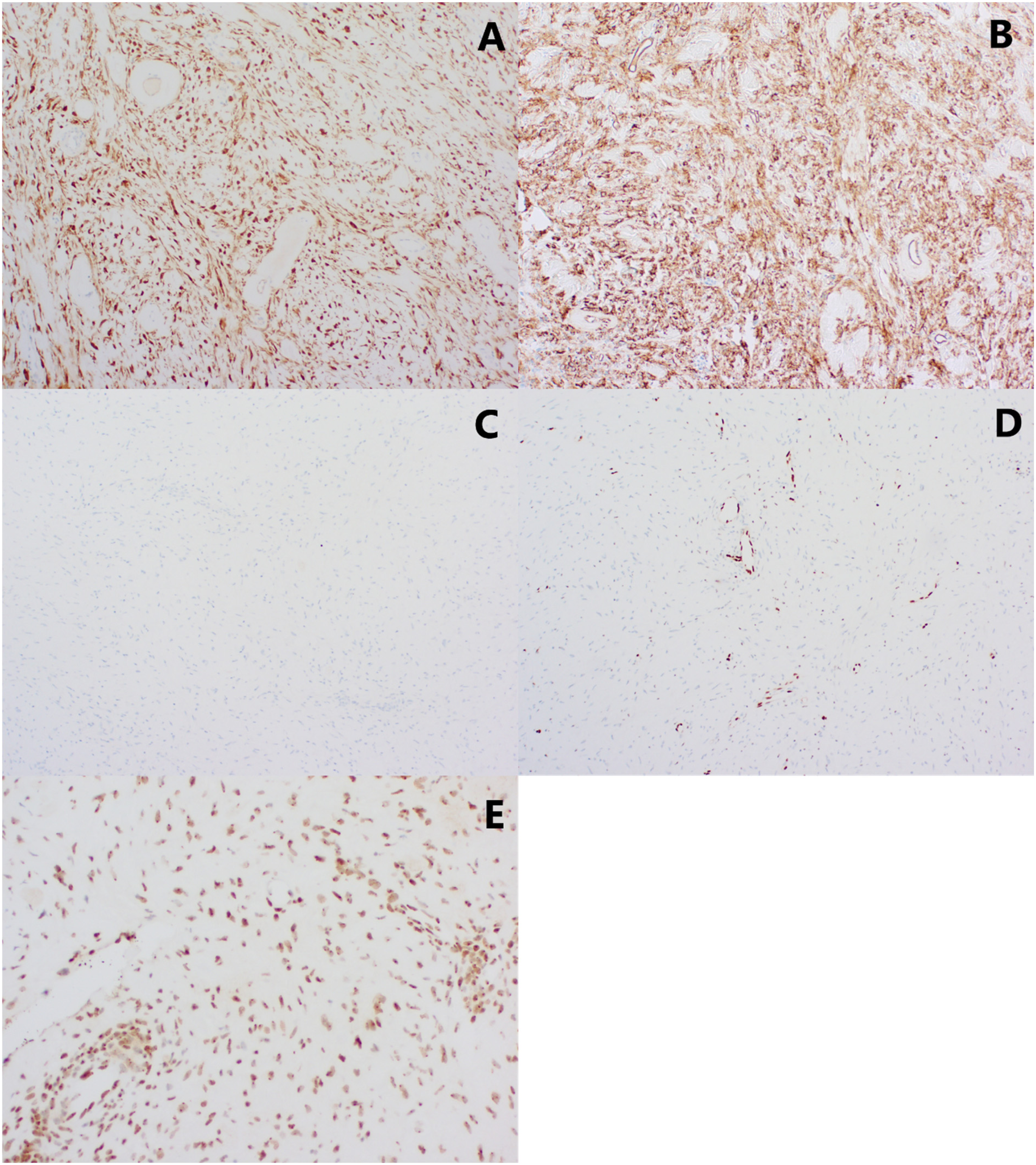
Immunohistochemistry on the spindle cells were positive for S100 (A) and CD34 (B). There was no expression of SOX10 (C) and ERG (D) in the spindle cells. Expression of H3K27me3 was retained in the tumor cells (E).
The morphological and immunohistochemical characteristics of this tumor were compatible with the recently described emerging group of molecularly defined rare spindle cell soft tissue tumors showing S100 and CD34 co-expression and harboring NTRK gene rearrangements or other gene alterations implicated in receptor tyrosine kinase pathway activation (such as BRAF, RET, or RAF1).
RNA-based Next-Generation Sequencing
RNA extraction from FFPE was performed using the Maxwell(R) RSC RNA FFPE kit. RNA-based next-generation sequencing with the Archer FusionPlex Expanded Sarcoma panel (55 genes) was carried out and revealed a SLMAP::RAF1 fusion, involving exon 9 of the SLMAP gene (NM_007159.4) and exon 8 of RAF1 gene (NM_002880.3) (Figure 4).
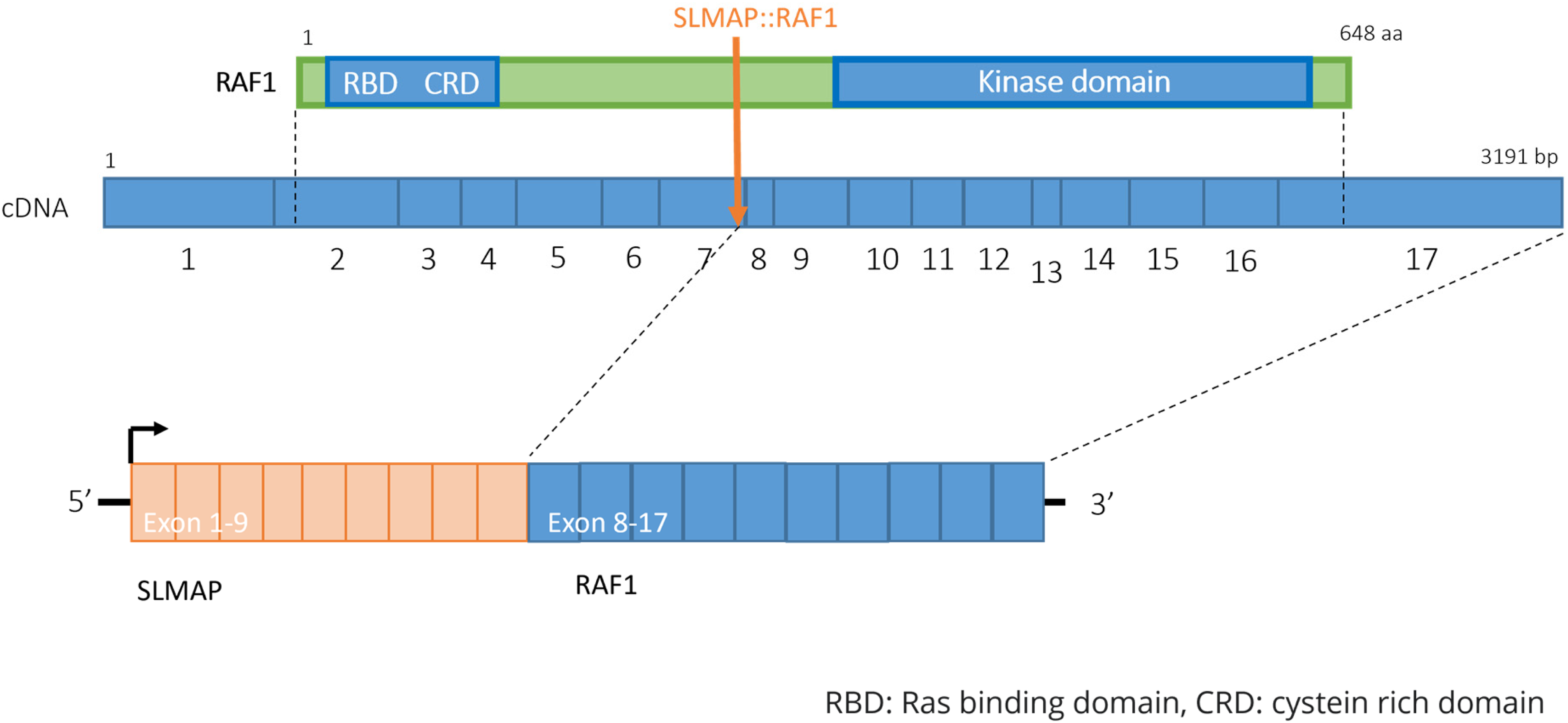
SLMAP::RAF1 gene fusion.
Treatment
Excision of the lesion was performed under general anesthesia and the abdominal wall was reconstructed with synthetic mesh. Pathological examination showed similar histology and immunohistochemistry as seen on the biopsy. Remarkably, there was calcification and heterotopic ossification (with positivity for SATB2) in the center of the lesion (Figure 5). The lesion was completely removed with tight margins. The patient experienced no complications and could resume her activities after 6 weeks. Sixteen months after the excision, there is no local recurrence on follow-up ultrasound.
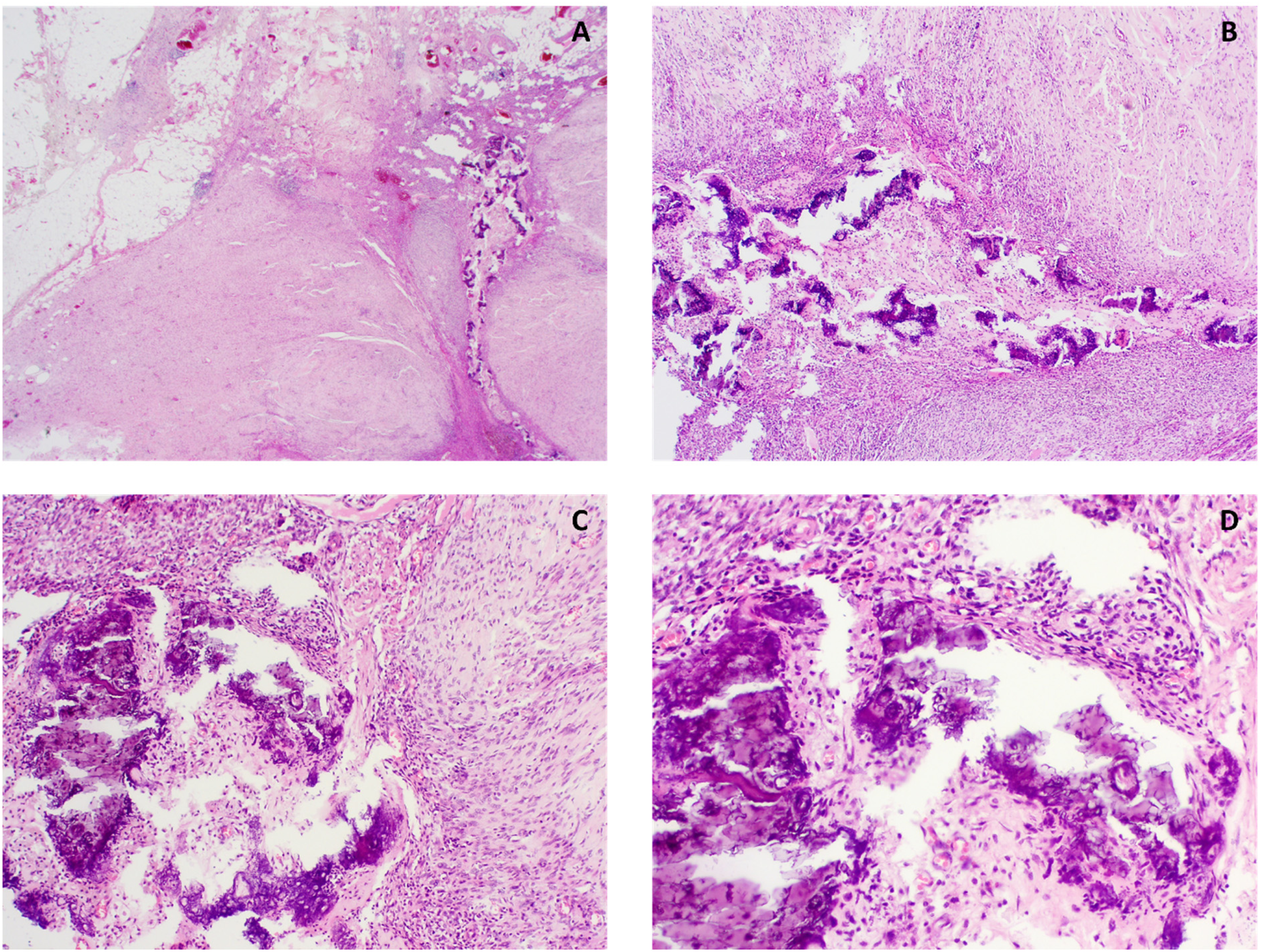
Calcifications and heterotopic osseous metaplasia in the center of the lesion ( Hematoxylin and eosin [HE] original magnification 20× (A), 40× (B), 100× (C) and 200× (D)).
Discussion
The group of spindle cell tumors with co-reactivity of S100 and CD34 and with NTRK gene rearrangements or other gene alterations involved in the activation of the receptor tyrosine kinase pathway, is most common in children and young adults (especially when the tumor contains an NTRK gene fusion) and occurs mainly in the extremities.2,3 They depend on molecular diagnostics for accurate classification as they belong to the group of fusion-positive spindle cell mesenchymal tumors.1–4
These tumors exhibit a wide spectrum of morphologies but are usually characterized by monomorphic spindle cells with a random pattern (patternless architecture). Focally pleomorphic or multinucleate cells can be seen. They have a range of low-grade (with low cellularity, low mitotic activity and monomorphic cytology) to high-grade features (with increased cellularity and increased mitotic activity, but no necrosis). 3
Additionally, three groups can be distinguished based on their morphology:
Lipofibromatosis-like morphology with an infiltrative growth pattern in adipose tissue. These tumors typically harbor a NTRK1 gene fusion. A smaller subset (typically occurring in children) show RET gene fusions. Solid growth pattern with low-grade morphology, stromal hyalinization and perivascular keloid-like hyalinized collagen, resembling malignant peripheral nerve sheath tumors (MPNST). Fibrosarcoma-like morphology (high-grade phenotype) with increased cellularity and mitotic activity, but no necrosis.1,2–8
Lipofibromatosis-like areas or the presence of pleomorphic/multinucleated cells have not been reported in cases with an RAF1 gene rearrangement.1,3,4 Tumors with a malignant phenotype (with increased cellularity and mitotic activity) have a malignant potential with proven metastasis, relapse, or death.1,2,3 Tumors with low cellularity show an indolent course.2,3 The described lesion belongs to the second group, with a morphology resembling an MPNST. However, all these lesions show co-expression of S100 and CD34, with negativity for SOX10 and retained expression for H3K27me3.1,2,5,9–11 This is in contrast with MPNST.
Molecular studies detected the presence of gene fusions with different kinases, mainly NTRK1, NTRK2, and BRAF, in these spindle cell tumors with S100 and CD34 co-reactivity. In addition, also RAF1, RET, and ALK gene rearrangements have been reported in tumors with the same morphological and immunohistochemical characteristics.5,9–11 Pan-TRK immunohistochemistry is positive in the tumors containing an NTRK fusion, but negative in the others.1–3
In our tumor, a SLMAP::RAF1 fusion was detected by RNA NGS. RAF1 is a member of the RAF family and part of the MEK-ERK signaling pathway that promotes cell proliferation, transformation and survival.1,3,12 RAF1 fusions have already been reported in carcinomas (prostate, breast, thyroid, and pancreas), melanomas, primary bone sarcomas, soft tissue sarcomas, pediatric (brain) tumors such as low-grade gliomas, glioblastomas, Langerhans-cell histiocytosis, desmoplastic infantile gangliogliomas and (anaplastic) pleomorphic xanthoastrocytomas.7,8,12–18 The presence of RAF1 gene fusions was first described in soft tissue tumors by Suurmeijer et al 3 in 2018, with PDZRN3, SLMAP, and TMF1 as its gene fusion partners. They found 8 patients with spindle cell tumors with S100 and CD34 co-reactivity harboring a RAF1 gene fusion/rearrangement. Of these 8 lesions, 6 occurred in adults, and only 1 harbored a SLMAP::RAF1 gene fusion. This fusion was detected by targeted RNA sequencing and occurred in a male adult with localization of the lesion in the thigh. Histologically, this lesion showed a low cellularity with a pure low-grade morphology, compatible with our case. The detected gene fusion in this lesion was also the same as seen in our tumor and was the result of an intrachromosomal inversion. SLMAP is fused to exon 8 of RAF1 gene (in both their and in our case), such that the fusion products contain the kinase domain of RAF1 (encoded by exons 10-17, Figure 4). 12 Recently, Zhang et al 4 described two lesions of spindle cell tumors with RAF1 rearrangement in children, including a newly described FMRA::RAF1 fusion.
To the best of our knowledge, calcifications or heterotopic ossifications in RAF1-rearranged spindle cell mesenchymal tumors have not yet been described in the literature. Only one patient with an infiltrative sarcoma with a PDZRN3::RAF1 gene fusion that showed focal areas of heterologous chondroid matrix deposition was published by Suurmeijer et al. 3 The presence of calcifications may be due to the long-term nature of the lesion or may be related to the morphology of the lesion, which resembles MPNST. Heterologous bone metaplasia and calcifications have already been described in neurogenic lesions, including neurofibromas, schwannomas and MPNST.2,19 More cases will be required to establish this difference.
RAF1 rearrangements have also been reported in an infantile fibrosarcoma (IFS). This is a fibroblastic neoplasm of soft tissue with fascicular or herringbone architecture, occurring in children. Typically, they show an ETV6::NTRK3 gene fusion. In one reported IFS with RAF1 rearrangement there was no reactivity for S100, CD34 or SOX10.1,6,20 These tumors show an underlying shared molecular pathway, but the relationship between IFS and spindle cell mesenchymal tumors with S100 and CD34 co-expression remains unclear.
A low threshold for molecular analysis should be maintained in spindle cell mesenchymal tumors with an overlapping morphology and nonspecific immunohistochemical profile since molecular markers aid in the classification and patients can have benefit from targetable therapies. 3 However, for some RAF1 gene fusions (ie QKI::RAF1 and SRGAP3::RAF1), it has been found that they activate both the MAPK and PI3 K/mTOR pathway and that first- and second-generation RAF inhibitors might be ineffective for RAF1 fusion inhibition. A combination therapy targeting MAPK and PI3 K/mTOR pathways or the use of pan-RAF dimer inhibitors are proposed. 21 Baranov et al 22 described a tyrosine-kinase-altered spindle cell tumor occurring in a 1-year-old boy showing a MAP4::RAF1 fusion (CD34 and S100 negative on immunohistochemistry). The patient was treated with trametinib (=MEK inhibitor that inhibits the mitogen-activated protein kinase enzymes MEK1 and/or MEK2 and thus affects the MAPK/ERK pathway) for 4,2 months and showed a partial response. Also, a patient with a metastatic melanoma with an AN010::RAF1 fusion showed a significant clinical response to MEK inhibition.18,23
In the diagnostic workup of a spindle cell, mesenchymal tumor with co-expression of CD34 and S100 a pan-TRK IHC should be performed for detection of an underlying NTRK fusion with further molecular analysis if pan-TRK IHC is positive (for molecular confirmation of the NTRK fusion) or negative (for further molecular classification). RNA NGS is the most widely used test for the detection of pathogenic gene fusions.1,6
Conclusion
In conclusion, we report an exceptional case of a spindle cell mesenchymal tumor with S100 and CD34 co-reactivity, which harbored a SLMAP::RAF1 fusion. Remarkable was also the presence of calcification and heterotopic ossification in the center of the lesion, a feature that, to our knowledge, has not been described yet in RAF1-rearranged spindle cell mesenchymal tumors. A low threshold for molecular analysis should be maintained in these spindle cell mesenchymal tumors and the pathological report should contain the clinically relevant molecular alterations since they can serve as a diagnostic marker and have been shown to be therapeutically targetable.1,6
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethical Approval
Ethical approval for this study was obtained from the ethical committee of the Ghent University Hospital (EC/077-2022/sds).
Informed Consent
Informed consent was obtained from the patient.