
Abstract
Background:
Defects in thyroid hormone synthesis at birth lead to congenital hypothyroidism (CH). Recently, pathogenic variants in the SLC26A7 gene have been linked to dyshormonogenetic goitrous CH. This anion transporter is highly expressed in the thyroid and is involved in thyroid hormone synthesis; however, its exact function and cellular localization remain unclear. In this study, we investigated SLC26A7 variants in Finnish patients with CH, characterized the phenotypes, and analyzed thyroid-specific gene expression.
Methods:
SLC26A7 variants were identified from a clinical CH cohort (n = 139) using exome sequencing, and the FinnGen database (R12 release) was screened for disease associations. Thyroid histology and thyroid-specific gene expression were analyzed in six human samples (including two homozygous SLC26A7 pathogenic variant carriers, patients with goitrous and hyperactive thyroids, and normal controls) and in thyroids from different mouse models (including hypo- and hyperthyroid mice, thyroid-specific G-protein deficient, and Slc26a7-knockout mice).
Results:
Four CH patients from four novel families carried the homozygous SLC26A7 (c.1893delT, p.F631Lfs*8) pathogenic variant. Two had large trachea-compressing goiters, requiring thyroidectomy already at birth. In addition, one homozygous participant with normal CH screening results developed hypothyroidism at age 16, and one patient with heterozygous SLC26A7 pathogenic variant had permanent CH at birth. Dentofacial abnormalities were frequently noted, including enamel hypoplasia (in four carriers), pro- or retrognathia, and malocclusion requiring orthodontic treatment (in 8/24 carriers). Thyrocyte hypertrophy with large colloid aggregates was a hallmark of homozygous patients. FinnGen screening revealed a 75-fold enrichment of the variant in the Finnish population, identifying a few other homozygous and seven heterozygous cases with early-onset hypothyroidism and dentofacial abnormalities. In human thyrocytes, SLC26A7 was localized to the basolateral membrane, with intense staining in hyperthyroid samples, while in mouse thyroid models, its expression pattern depended on dietary iodide levels, thyrotropin signaling, and GNAS activity.
Conclusions:
We describe variable phenotypes associated with the SLC26A7 pathogenic variant, ranging from severe CH with large congenital goiters to delayed onset hypothyroidism and dentofacial abnormalities. SLC26A7 shows thyrotropin-, GNAS-, and dietary iodine-dependent basolateral localization, suggesting their role in phenotypic variations.
Keywords
Introduction
Lack of thyroid hormones at birth leads to congenital hypothyroidism (CH), affecting 1 in 1000 to 1 in 4000 newborns worldwide, with an incidence of 1:2783 in Finland.1–3 CH is typically a sporadic disease with unknown etiology, but ∼10–20% of cases are familial and caused by pathogenic variants in genes involved in thyroid development or hormone synthesis.4,5 Despite advances in high-throughput sequencing, a proportion of familial cases remain unexplained.
Most CH cases result from thyroid dysgenesis: aplasia, hypoplasia, or ectopy. 6 However, genetic variants are identified in only a minority (2–5%) of these cases.5,7 In contrast, thyroid dyshormonogenesis has a more substantial genetic background, with pathogenic variants identified in over half of the cases, particularly in familial goitrous CH.5,8 Previously reported causative genes include the sodium iodide symporter (NIS, SLC5A5), apical iodide transporter pendrin (SLC26A4), thyroid peroxidase (TPO), thyroglobulin (TG), and dual oxidases (DUOX2 and DUOXA2). 9
A more recently implicated gene in thyroid dyshormonogenesis is SLC26A7, a member of the solute carrier family 26. SLC family transporters are essential for thyroid hormone synthesis. SLC5A5 mediates iodide uptake, and SLC26A4 transports iodide into the colloid. However, pendrin’s role as the primary apical iodide transporter is debated due to the identification of additional transporters. 10
SLC26A7 was initially cloned from the human kidney as an anion exchanger, with expression also found in the placenta, testis, and retina, but not yet studied in the thyroid.11–13 High expression in the thyroid was later identified, and homozygous inactivating pathogenic variants were discovered in patients with goitrous CH and normal renal function, including three Finnish families revisited here.14,15 Thereafter, a few additional families with homozygous SLC26A7 pathogenic variants and CH have been reported.14–18 Patients typically present with goitrous CH, impaired iodine organification, and normal or mildly elevated iodide uptake.
Slc26a7-null mice develop goiter and hypothyroidism,14,19 but unlike humans, they also show renal tubular acidosis and reduced thyroidal iodide uptake.14,20 High-iodine diets partially rescue the hypothyroid phenotype. 14 Dietary iodine serves as the essential substrate for thyroid hormone synthesis, and sufficient iodine intake can partially compensate for impaired iodide transport or other defects in hormone production. Thus, the expression of SLC26A7 differs between humans and mice. Initially described as a Cl−/HCO3− exchanger, SLC26A7 was proposed to regulate pH and ionic balance for thyroid hormone synthesis enzymes (TPO, DUOX2). Later, a dual-compartment cell system indicated that SLC26A7 also functions as an apical iodide transporter. 15 Thus, its precise role in thyroid hormone synthesis remained unresolved.
To gain insight into the phenotypic and functional significance of SLC26A7, we screened this pathogenic variant in a Finnish cohort of CH patients, characterized their phenotypes and thyroid histology, and examined SLC26A7 expression in thyroid disease models. We also explored disease association in the FinnGen database. 21
Materials and Methods
We screened 139 Finnish CH patients for SLC26A7 variants using exome sequencing, with confirmation by Sanger sequencing. Of these, 51 patients were identified through hospital registries in the catchment areas of Turku and Kuopio University Hospitals and provided blood samples for CH-related genetic testing. This part of the study was approved by the Ethics Committee of Northern Savo Hospital District (March 13, 2018; No 346/2018). The remaining 88 patients were recruited by pediatric endocrinologists across Finland and also provided blood samples for genetic testing. This part of the study was approved by the Ethics Committee of the Hospital District of Southwest Finland (108/180/2010).
CH was defined as abnormal thyroid function tests (TFTs) in newborn screening, except patient Ag, who was born before the screening era. She had obvious CH-related symptoms and started on levothyroxine (LT4) at the age of one year. In Finland, CH screening is based on umbilical cord thyrotropin (uS-TSH). If uS-TSH exceeds 40 mU/L, a confirmatory serum sample is collected at approximately 3 days of age to measure both thyrotropin (TSH) and free thyroxine (fT4) levels using standard assays in local hospital laboratories. A TSH level > 20 mU/L together with a fT4 < 10 pmol/L is generally considered diagnostic for CH, although reference ranges have varied slightly over the decades depending on the laboratory as described more in detail in the Supplementary Data.
Phenotypic data were obtained through interviews and manual review of medical records, including the course of hypothyroidism, TFTs, and other diagnoses. Written informed consent was obtained from all participants or their legal guardians.
This study was approved by the Ethics Committee of the Hospital District of Southwest Finland (108/180/2010) and conducted following the Declaration of Helsinki (2024). A detailed methodology is provided in the Supplementary Data.
Genetic analysis
DNA was isolated from peripheral blood using Qiagen kit (Valencia, CA) and used for either targeted-next generation sequencing or whole exome sequencing for index patients.22,23 Genomic variant analysis platform (Nostos Genomics GmbH) was used for identifying rare pathogenic/likely-pathogenic variants (MAF ≤ 0.01), based on ACMG/AMP 2015 guidelines and/or Clinvar database.24,25 Copy number variation (CNV) analysis was done for patient Da and Db using the whole exome data with the same abovementioned platform (AION v3.12.0.1, Nostos Genomics GmbH).
The SLC26A7_c.1886_1887delT variant identified in the index patients was validated and further screened in the affected and unaffected family members and other unrelated CH patients from our cohort (n = 139), including families previously described. 22 Sanger sequencing was done in Eurofins Genomics GATC Biotech (Germany). Alamut Visual Plus v1.9 software (SOPHiA GENETICS© 2023) was used to analyze the Sanger sequences. In this study, we focused specifically on characterizing patients carrying the SLC26A7 variant, while other genetic causes of CH identified in the cohort are reported separately.
Study approvals
The genetic screening of patient samples was approved by the Ethics Committee of the Northern Savo Hospital District (March 13, 2018; No. 346/2018) and by the Ethics Committee of the Hospital District of Southwest Finland (108/180/2010). Human thyroid FFPE samples were obtained from Departments of Pathology or Biobanks from University Hospitals in Finland, which have been collecting them according to ethics recommendations from the Helsinki declaration (2024). Normal human thyroid sections were purchased from Novus Biologicals (Novus Biologicals, USA).
All mouse FFPE thyroids were from the stored collections. Slc26a7-KO mice FFPE samples were a gift from N.S. The sections came from mice bred at the University of Cambridge. Mouse studies were regulated under the Animals (Scientific Procedures) Act 1986 Amendment Regulations 2012 following ethical review by the University of Cambridge Animal Welfare and Ethical Review Body (UK Home Office License no. P0101ED1D).
TSHR M453T-KI and TSHR-KO mice and goiter experiments were authorized by the National Animal Experiment Board of Finland with license no. 35039 26 and TSHR D633H-KI mice with license no. 10266. 27 Thyroid-specific Gαs-, Gαq/Gα11-, and Gα12/13-deficient mice had been approved by the Regierungspräsidium Karlsruhe (Karlsruhe, Germany). 28
Immunohistochemistry
FFPE-thyroid samples were processed according to standard immunohistochemistry (IHC) protocols, using Epitope retrieval solutions pH 6.0 or 9.0 (Leica) for antigen retrieval, and normal antibody diluent (ImmunoLogic a WellMed Company, Netherlands) with H2O2 for blocking. Staining was done with semiautomated Lab Vision autostainer (Thermo-Fisher Scientific). Human SLC26A7 was detected with rabbit polyclonal antibodies PA5-68485 (discontinued; Invitrogen) and CABT-L650R (Creative Diagnostics, USA), SLC5A5 with 14 F-mouse monoclonal antibody (LSBio, USA), SLC26A4 with rabbit polyclonal antibodies NBP1-85237 (discontinued; Novus Biologicals, USA) and bs-6787R (Bioss, USA), and thyroglobulin with mouse SPM221-monoclonal antibody (NeoBiotechnologies, USA). Mouse Slc26a7 was examined with mouse 14H5-monoclonal antibody (Santa Cruz Biotechnology, Inc.) and Slc5a5 with mouse G-5-monoclonal antibody (Santa Cruz Biotechnology, Inc.). Secondary antibodies were either Dako EnVision®+ Dual Link System-HRP (DAB+) (Agilent) or BrightVision 1 step detection system goat anti-mouse or anti-rabbit (ImmunoLogic a WellMed Company). Development was done with BrightDAB (ImmunoLogic a WellMed Company) and counterstaining with Gill No. 1 (Sigma-Aldrich) or Mayer’s (Reagena, Finland) hematoxylin. The slides were mounted with Pertex and imaged by Pannoramic 250 scanner (3DHISTECH Ltd, Hungary). To validate antibody specificity and reliable detection, we compared staining patterns between homozygous human SLC26A7 variant carrier thyroid samples, Slc26a7-knockout mouse tissue (which served as negative controls), and wild-type (WT) controls.
RNA sequencing
Slc26a7, Nis, pendrin, and thyroglobulin mRNA expression was studied from thyroids of homozygous Tshr M453T and WT mice, fed with either high-iodide (6 mg/kg) or sufficient-iodide (SI: 0.3 mg/kg) for 2 months (n = 3–8/group). RNA was isolated using NucleoSpin RNA isolation Kit (MACHEREY-NAGEL GmbH & Co.) and sequenced by Novogene Co. Ltd. in United Kingdom.
FinnGen analysis
Genotyping, QC, and imputation were performed as described in detail in the FinnGen resource article. 21 In FinnGen R12 (available at https://www.finngen.fi/en/access_results and https://r12.finngen.fi), Regenie version 2.2.4 was used with sex, age, 10 principal components of ancestry, and genotyping batch included as covariates. Recessive model was run for all protein-coding variants in FinnGen. Exact definitions of ICD [8,9,10] diagnoses, medications, and procedures for all FinnGen analyses are publicly available at https://risteys.finregistry.fi/ To test for significance and calculate OR, Fisher’s exact test from R 4.1.3 was used.
Statistical analysis
RNA-sequencing results were analyzed by three-way ANOVA with Tukey’s multiple comparisons test (significance at p ≤ 0.05). Statistics and figures were generated using the R Statistical Software version 4.1.2 for Windows, R Core Team 2021 (Vienna, Austria).
Results
Genetic, clinical, and biochemical characterization of families with CH
Following our previous report, 14 which identified the SLC26A7 c.1893delT pathogenic variant in three Finnish families with CH, here we screened for this SLC26A7 variant in an additional 139 patients with CH. We identified four novel unrelated families (Families A to D in Fig. 1) carrying the previously reported single-nucleotide deletion c.1893delT (p.F631Lfs*) in SLC26A7 (Fig. 1 and Supplementary Fig. S1). This frameshift variant results in a truncated protein within the STAS (antisigma factor antagonist) domain, impairing membrane localization and function, as described previously 14 and shown in Supplementary Fig. S2.
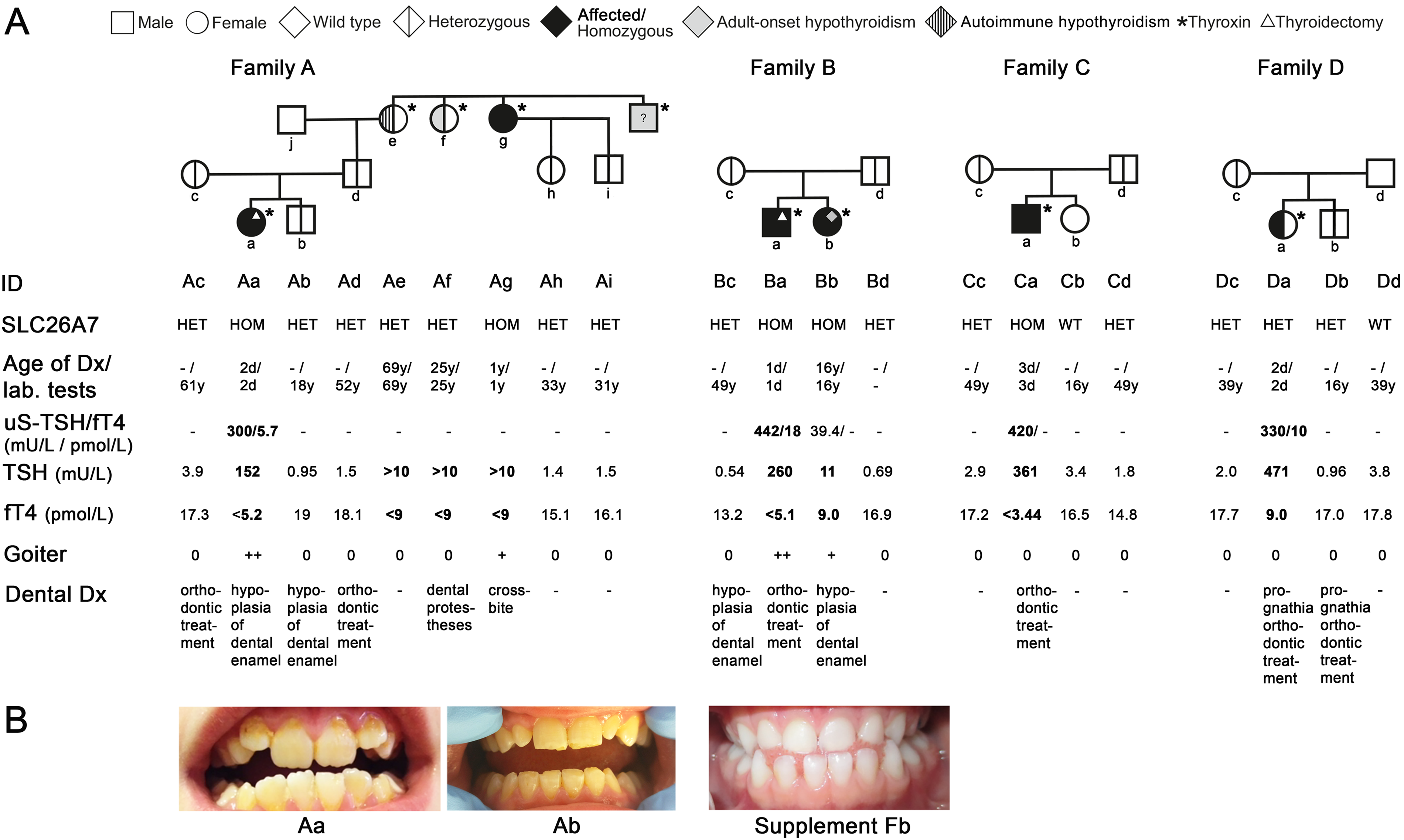
Modified pedigrees and clinical, biochemical, and genetic characterization of families with congenital hypothyroidism (CH) and SLC26A7 mutation.
Among these families, four CH patients were homozygous (Aa, Ag, Ba, Ca) and one (Da) was heterozygous for the pathogenic variant. In addition, one homozygous carrier (Bb) had normal thyroid function at birth, but developed nonautoimmune hypothyroidism at age 16, with otherwise normal development. All CH cases identified through neonatal screening began LT4 within 1–3 days of birth (Fig. 1). No other pathogenic variants were identified in these patients in coding regions of CH-related genes. 30 CNV analysis from exome sequencing data for the CH patient with a heterozygous variant (Da) did not reveal any pathogenic variation.
The index patients Aa, Ba, Ca, and Da had markedly elevated uS-TSH concentrations (range: 300–442 mU/L; screening threshold < 40 mU/L). Confirmatory testing showed TSH > 150 mU/L (threshold < 11 mU/L) and free T4 (fT4) < 5.3 pmol/L (normal range 11.5–28.3 pmol/L) in homozygous patients (Aa, Ba, Ca), consistent with severe CH. The heterozygous CH patient (Da) had TSH of 471 mU/L and fT4 of 9 pmol/L. A detailed summary of TSH, fT4, T3, and TG values is shown in Supplementary Table S1 and Supplementary Fig. S3. Patient Ca was initially considered to have transient CH, with normal thyroid function at 2.5 years of age and no LT4 therapy for five years. However, at the age of seven years, he developed hypothyroid symptoms, and TFTs confirmed severe nonautoimmune hypothyroidism (TSH 510 mIU/L, fT4 < 1 pmol/L, negative TPO autoantibodies). Notably, he never developed a goiter, and thyroid ultrasound findings were normal.
Family history in Family A revealed a paternal great-aunt (Ag) with CH, diagnosed at around one year of age before the start of neonatal screening. She had normal development, but developed a goiter during pregnancy.
In total, 5 of 24 heterozygous carriers across the newly and previously described families developed adolescent- or adult-onset hypothyroidism (three nonautoimmune, two autoimmune; Fig. 1, Supplementary Fig. S4). Two euthyroid heterozygous parents (Ac and Cd) had elevated TPO autoantibodies.
Overall, 5 of 10 homozygous CH patients developed goiters and 2 required thyroidectomy at birth.
Other medical conditions and dentofacial abnormalities
Other medical conditions were assessed through interviews, questionnaires, and a review of patient records. Dentofacial abnormalities were common, including enamel hypoplasia in two heterozygotes and two homozygous carriers as well as severe prognathia or other malocclusions requiring orthodontic treatment in four heterozygotes and four homozygous carriers. All patients exhibited normal growth and developmental milestones. No other significant health issues were observed, including infertility (all heterozygous families had multiple children, and one homozygous carrier had two children), abnormal growth, or other clinical symptoms (Fig. 1 and Supplementary Fig. S4).
Association study between SLC26A7 variants and phenotypes using FinnGen data
The SLC26A7 frameshift variant (8–91393989-AT-A, rs762022055 or rs774652174, p.Phe631LeufsTer8) is 75-fold enriched in the Finnish population and concentrated in the Northern Savonia region in the FinnGen data. We leveraged the FinnGen R12 cohort (n = 520,210) to comprehensively evaluate the impact of the SLC26A7 frameshift variant across the Finnish population. This cohort combines nationwide health care registry data with genomic information, allowing us to examine lifelong clinical outcomes linked to genetic variants. To account for health registry-based ascertainment that captures a spectrum of thyroid dysfunction beyond strictly congenital presentations, we defined early-onset hypothyroidism as diagnosis before the age of 13 years.
The SLC26A7 frameshift variant demonstrated complete penetrance for early hypothyroidism in homozygotes (p = 1.15 × 10−8, Fisher’s exact test). An association with sensorineural hearing loss before 47 years of age was also observed in homozygotes (OR = 98.7 [CI: 8.9–1,094.2], p = 0.0016). Furthermore, heterozygotes showed modestly increased risk of early hypothyroidism compared with noncarriers (OR = 2.42 [CI: 1.15–5.09], p = 0.0167). We examined birth records to confirm that FinnGen homozygotes were not members of the aforementioned clinical cohort families.
Notably, the extensive longitudinal clinical data in the FinnGen resource showed a striking association between the frameshift and concurrent presentation of early hypothyroidism with craniofacial features (retrognathia, ICD10 K017.11, and/or deep bite, ICD10 K07.23). While early-onset hypothyroidism occurs at a rate of 0.089% and retrognathia/deep bite at 1.08% in the FinnGen cohort, their co-occurrence was markedly enriched in variant carriers. A majority of homozygotes presented with both phenotypes (OR = 4851.3 [CI: 435.2–54,089.7], p = 2.89 × 10−9), and heterozygotes showed a significantly elevated rate of concurrent presentation (OR = 147.3, [CI: 46.8–463.5], p = 1.73 × 10−5).
In addition, FinnGen data show a significant association between this frameshift variant and tooth agenesis (p = 1.29 × 10−6, OR = 2676) in the whole cohort, not specific to early-onset hypothyroidism. The biological significance of these findings has been further validated by a recent single-cell RNA sequencing analysis of rat mandibular molars, which demonstrated that Slc26a7 is specifically expressed during dental follicle cell differentiation. 31 This suggests that SLC26A7 plays a crucial role in both thyroid function and craniofacial development, with particularly pronounced effects in individuals who develop early hypothyroidism. Beyond these phenotypes, no other significant associations with this frameshift variant were identified in a comprehensive scan of over 2200 FinnGen phenotypes.
Thyroid IHC in SLC26A7 mutant carriers and euthyroid, hyperthyroid, and goiter patients
IHC analyses of thyroid samples from homozygous SLC26A7 p.F631Lfs* variant carriers (index cases Aa, Ba) revealed features of dyshormonogenetic goiter, including thyrocyte hyperplasia and hypertrophy with large colloid aggregates, which appear to be characteristics of this condition, and no basolateral SLC26A7 staining (Fig. 2a,b). Intense basolateral staining was present in goiter and hyperthyroid samples and also detectable in euthyroid thyroid tissue (Fig. 2). As evaluated also with antibody used in mouse studies, we found weak immunoreactivity also close to nucleus in all human samples (Supplementary Fig. S8). Notably, SLC26A7 showed consistent basolateral membrane localization in both human and mouse thyrocytes, which contrasts with some previous reports suggesting apical localization. 17 To assess the impact of the variant on thyroid protein expression, we analyzed NIS, pendrin, and thyroglobulin by IHC (Fig. 2). Pronounced basolateral NIS staining was present in the thyroid of patient Ba (thyroidectomy at birth prior LT4 treatment), while minimal staining was observed in patient Aa (thyroidectomy at day 10, after eight days of LT4). In the goiter control sample (unknown etiology), NIS expression was also low, whereas abundant but more variable basolateral NIS staining was detected in the hyperthyroid sample. Pendrin showed consistent apical localization across all samples. Thyroglobulin staining was present in all samples, but intense in patients Aa and Ba. Furthermore, both thyroglobulin and hematoxylin–eosin staining detected numerous white aggregates in colloid in these patients and less in goiter patient. Additional thyroid markers, examined earlier in hyperthyroid patient carrying TSHR D633H-variant, are shown in Supplementary Fig. S5A. In summary, homozygous SLC26A7 mutants exhibited characteristic thyrocyte hypertrophy, colloid densification, and differential NIS regulation, while pendrin expression remained preserved.
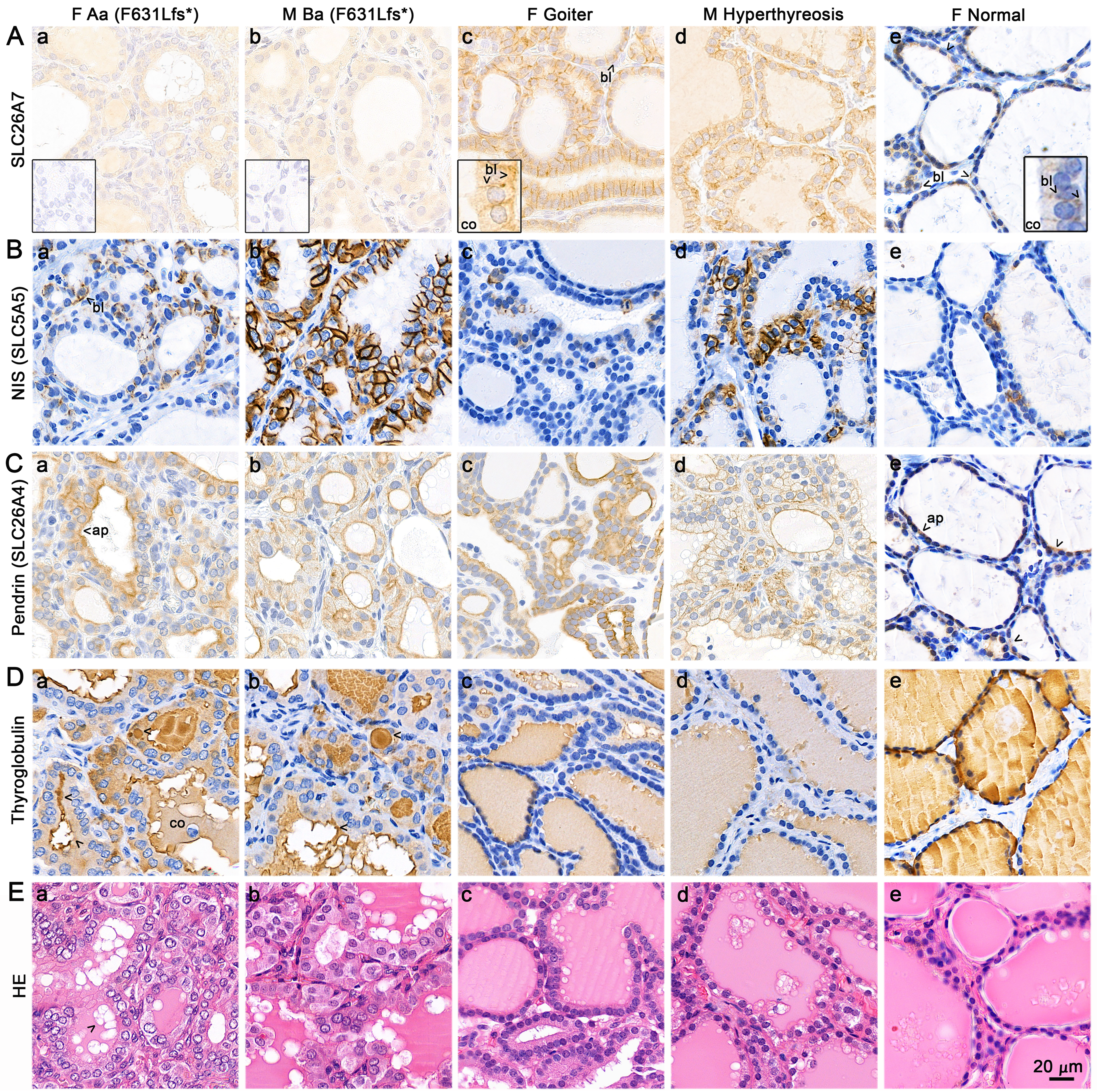
Immunohistochemistry of human thyroid samples from two homozygous SLC26A7 p.F631Lfs*carriers and goiter, hyperthyroid, and control tissues.
TSHR-dependent regulation of SLC26A7 localization in mouse models
Having established the consequences of SLC26A7 deficiency in human thyroid tissue, we next investigated mechanisms controlling SLC26A7 expression using variety of mouse models, including Slc26a7-knockout (KO), TSH-injection and goiter models, TSH-receptor KO (Tshr-KO) model, Tshr knock-in (KI) models (constitutively active Tshr-M453T-KI 26 and Tshr-D633H-KI 27 ) as well as thyroid-specific Gαs-, Gαq/11-, and Gα12/13-deficient models.28,32,33 Previous in vitro studies have shown that the TSH-TSHR pathway regulates SLC26A7, although the regulation appears to occur at the level of protein translocation to the cell membrane rather than transcriptional upregulation. 34 Therefore, we used these in vivo models to investigate whether SLC26A7 expression and localization are dependent on the TSH receptor pathway and its downstream G-protein-coupled signaling cascades.
A validated commercial antibody showed intense basolateral Slc26a7 staining in WT thyrocytes and no staining in Slc26a7-null thyroids (Fig. 3Aa,b), but pronounced staining with Nis (Fig. 3Ba), resembling human index cases Aa and Ba. The thyroid morphology also resembled index cases Aa and Ba. Abundant SLC26A7 staining was observed in WT mice following TSH injection or after exposure to goitrogenic diet, compared with controls (Fig. 3Ac–f). TSH injection led to a slight increase in NIS expression, whereas goitrogen treatment produced a more pronounced effect (Fig. 3Bc–f). Tshr-KO mice exhibited weak SLC26A7 and NIS staining in residual follicles (Fig. 3Ag, Bg). In Tshr-KI mice, abundant dietary iodine-dependent SLC26A7 membrane staining was detected (Fig. 4A, Supplementary Fig. S5B). Nis was mainly present in Tshr-M453T-KI females, fed with SI diet (Supplementary Fig. 7e). However, RNA sequencing of whole thyroids from Tshr-M453T-KI and WT littermates showed no significant difference in Slc26a7 mRNA levels, but pendrin and Nis transcripts instead were significantly increased in Tshr-M453T-KI mice under sufficient iodide intake (Fig. 4B).
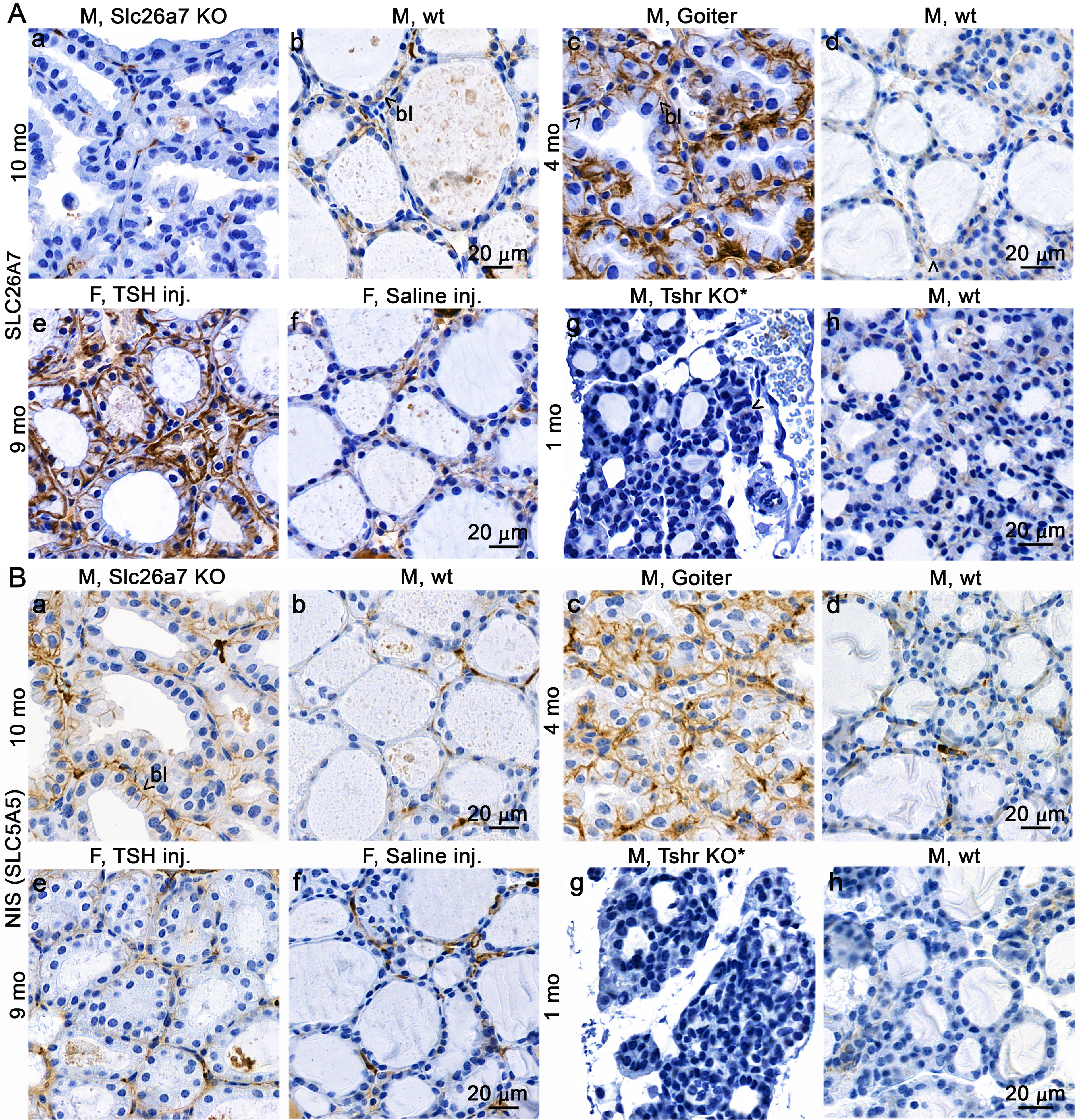
Slc26a7 and Nis expression and localization in thyroids of various mouse models.
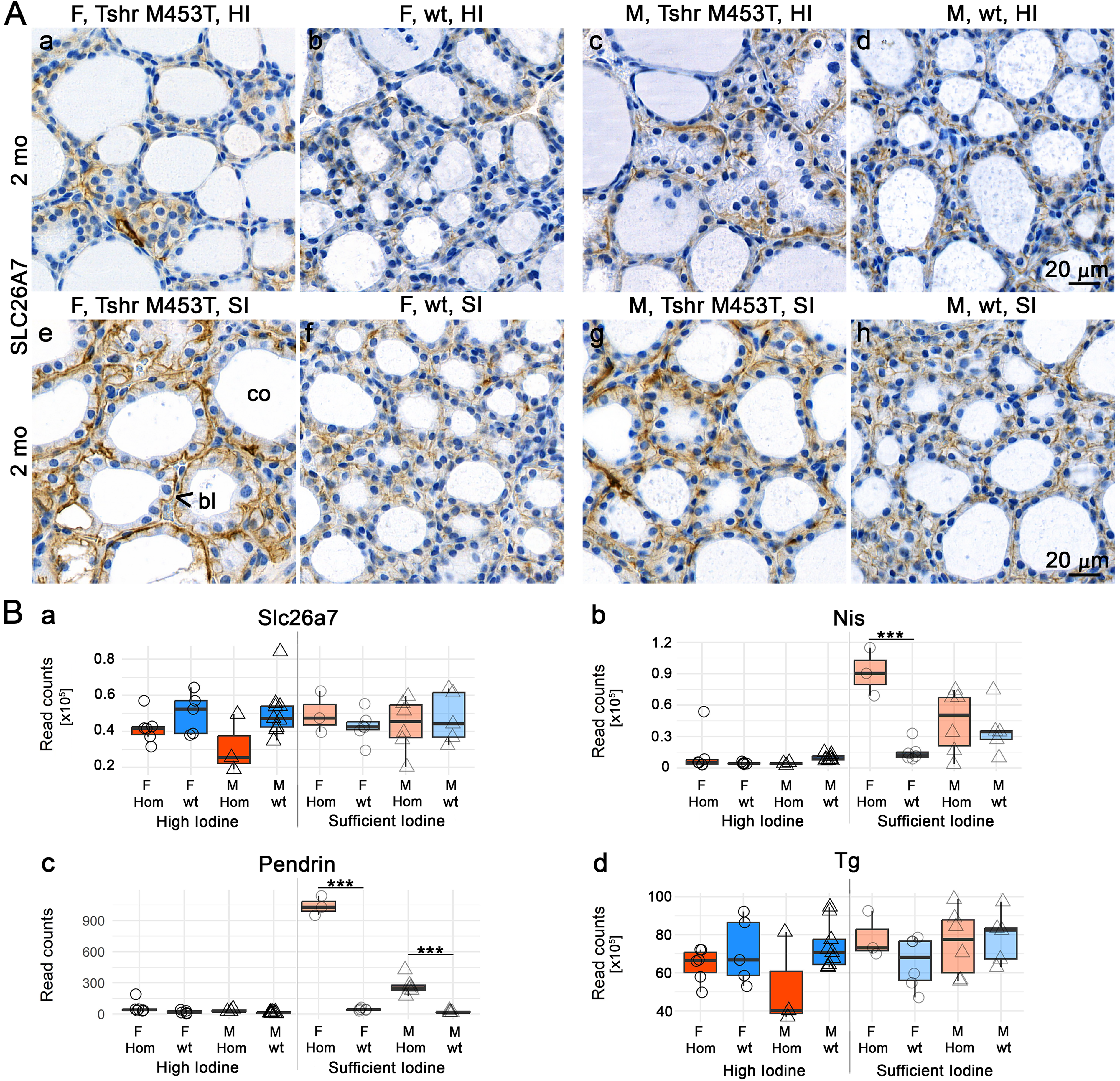
Slc26a7 immunostaining and mRNA expression in relation to dietary iodide in the nonautoimmune hyperthyroid Tshr-M453T knock-in (KI) mouse model. Mice were fed a normal, high-iodide (HI, 6 mg/kg), or sufficient-iodide (SI, 0.3 mg/kg) diet for 2 months.
As in Tshr-M453T-KI mice, the variant leads mainly Gαs-PKA 26 activation, whereas in Tshr-D633H-KI model also Gαq/11-PKC 27 -mediated signaling is activated; our effort was to clarify the downstream pathways that lead to translocation of SLC26A7 to the cell membrane. Therefore, we studied the expression of Slc26a7 also in different thyroid-specific G-protein alpha subunit (Gαs, Gαq/11, or Gα12/13) deficient thyroids.28,32,33 SLC26A7 expression was markedly reduced in Gαs-deficient thyrocytes, but remained unchanged in Gαq/11 and Gα12/13-deficiency models (Supplementary Fig. S6). Collectively, these findings indicate that TSH primarily affects SLC26A7 expression in cell membranes through Gαs-mediated cAMP/PKA-signaling, but not through Gαq/11 or G12/13 pathways.
Discussion
Pathogenic variants in SLC26A7 have recently been identified as a rare cause of familial dyshormonogenetic CH, although its precise role in thyroid hormone synthesis remains debated, particularly regarding its localization and regulation. The basolateral localization we observed differs from some reports suggesting apical localization of SLC26A7. 17 This discrepancy may reflect differences in antibody specificity, as we carefully validated our antibodies using knockout controls and variant carrier samples. It is also possible that different SLC26 family members with sequence similarities were detected in previous studies. To date, goitrous CH has been reported in approximately 10 families with SLC26A7 variants worldwide. We demonstrate that the SLC26A7 c.1893delT (p.Phe631LeufsTer8) variant is highly enriched in Finland and represents one of the most common genetic causes of familial CH in Finland, present in 7 of 25 screened familial cases from the 139 CH patients. Its carrier frequency is ∼1 in 150; therefore, alongside TPO pathogenic variants, 22 it is among the most common genetic causes of familial CH in Finland. All affected individuals in this study carried the same pathogenic variant, resulting in a frameshift and early truncation near the C-terminus within the STAS-domain more distal than previously reported variants in other countries (Supplementary Fig. S2). The founder effect is a possible explanation for this, due to the isolated nature and bottleneck phenomenon of the Finnish population. 21
Among our newly identified families and extended pedigrees, goitrous CH was observed in 5/10 homozygous CH patients, severe CH at screening in 10/11 homozygous patients, and late-onset hypothyroidism in one homozygous patient. Nongoitrous CH occurred in one heterozygous patient, while adult-onset nonautoimmune hypothyroidism developed in three heterozygous carriers and two developed autoimmune hypothyroidism. Dentofacial abnormalities, including deep bite, tooth agenesis, and jaw anomalies, were frequently observed and corroborated by FinnGen data. No other significant diagnoses were observed in carriers of the frameshift variant.
Characterization of SLC26A7 expression in patients and thyroid disease models revealed that SLC26A7 localizes to the basolateral membrane on thyrocytes. TSH stimulates its membrane localization via Gαs signaling, and its membrane expression is further influenced by dietary iodine suggesting that these factors could contribute to the phenotypic variability seen among patients with SLC26A7 variants.
Variability of CH and goiter phenotypes among SLC26A7 variant carriers
Insufficient iodine intake during pregnancy is a known cause of elevated TSH and neonatal goiter. 35 In Finland, 70% of mothers have been reported to have suboptimal iodine intake during late pregnancy.36,37 This may partly explain the phenotypic variation observed among our patients, although we lack maternal iodine data. A recent case report showed that 1 mg of iodine supplementation daily failed to normalize thyroid function in a SLC26A7-variant carrying individual. 18 Goiter variability is common in dyshormonogenesis and may be influenced by iodine status, genetic modifiers, or environmental exposures such as goitrogens. 4
A limitation of our study is that patients did not undergo perchlorate discharge tests or radioactive iodine uptake testing, as these are not routinely performed in Finland. Such tests would have provided additional insight into thyroid function in variant carriers.
We did not identify additional coding variants in known CH genes that could explain the phenotypic differences. The heterozygous variant in patient Da is unlikely to cause CH on its own, but it may confer an increased risk in the presence of other factors. Possible explanations for atypical presentations in heterozygotes include undetected large intronic or intergenic deletions, insertions, or duplications, deep intronic variants affecting regulatory elements, somatic variants, environmental influences, or post-translational changes. Future whole-genome sequencing may help to clarify these possibilities. Although symptomatic heterozygotes in recessive diseases are rare, similar cases have been reported in CH38–40 and other genetic disorders.41–46 Oliver-Petit et al. showed that biallelic and oligogenic variants are associated with severe CH and goiter, while heterozygous variants in TG, TPO, DUOX2, and TSHR are more often linked to mild or moderate CH with lower goiter risk. 37 According to European Society for Paediatric Endocrinology criteria for CH severity, 8 patient Da would be classified as moderate (fT4 < 10 pmol/L). In another study, Fugazzola et al. described affected heterozygotes with autosomal recessive CH caused by monoallelic expression of a mutant TPO allele. 39
Dentofacial abnormalities in SLC26A7 patients
In our cohort, 12/24 individuals with SLC26A7 pathogenic variant reported dentofacial abnormalities, such as deep bite and jaw misalignment occurring more frequently than in the general population (Fig. 1; Supplementary Fig. S4). This suggests a role for SLC26A7 in craniofacial development.
In mice, double KO of Slc26a7 and Slc26a1 delays enamel maturation, consistent with their proposed function as pH-regulating ion transporters during enamel maturation. 47 Dentofacial and skeletal abnormalities have also been described in other SLC family members, sharing mechanisms related to mineralization.48,49
SLC26A7 was originally characterized as a chloride-base exchanger on the basolateral membrane of acid-secreting cells in the renal outer medullary collecting duct and gastric parietal cells. 11 Slc26a7-deficient KO-mice show renal acidosis and impaired gastric acid secretion. 20 Later, in high-throughput screening of skeletal phenotypes in KO mice, these mice showed slightly reduced lean body mass, shortened femur length, increased trabecular bone mineral density, and hypothyroidism. 19
In our cohort, no major health issues beyond hypothyroidism and dentofacial abnormalities were noted, although one homozygous and two heterozygous carriers had a diagnosis of gastroesophageal reflux, indicating that the variant does not prevent this condition, despite reduced gastric acid secretion. 20
Mechanisms and possible function of SLC26A7 in thyroid
The role of SLC26A7 in iodine transport and thyroid physiology remains an area of ongoing investigation. In KO-mouse models, absence of Slc26a7 leads to goiter and hypothyroidism. 14 In vitro studies using polarized cells suggest that Slc26a7 can mediate iodide transport. 17
Here we used human samples (SLC26A7 variant carriers, goiter, and hyperthyroidism) and different mouse models to explore the histology and regulation of SLC26A7 expression under varying thyroid conditions (hypo-hyperthyroidism, TSH injection, and goitrogenic diet) and to specifically assess the role of TSHR and downstream pathways. Histological analysis of thyroidectomy samples from two patients with large goiter revealed features of dyshormonogenetic goiter, including thyrocyte hyperplasia and hypertrophy with large colloid aggregates, which appear to be characteristics of this condition. IHC revealed abundant SLC26A7 membrane expression in thyroid of human patients with hyperthyroidism compared with euthyroid sample. Thus, our findings suggest that TSH, Gαs signaling, and iodine status regulate the expression of SLC26A7 in the basolateral membrane. Supported by both human thyroid tissue and mouse models, TSHR activation through activating variants, TSH injection, or elevation using goitrogenic diet induced membrane expression in a cAMP-dependent manner, as previously shown in vitro. 40 These results, although based mainly on IHC, support a role for SLC26A7 in anion transport on the basolateral membrane, rather than as a primary apical iodide transporter. 15 Our basolateral localization finding does not necessarily contradict the observed organification defects in patients. 14 As we used commercial species-specific antibodies validated with SLC26A7 knockout controls, we are confident in the basolateral localization. The organification defect likely results from the possible role of SLC26A7 in maintaining proper pH homeostasis within thyrocytes and colloid through its chloride-bicarbonate exchanger activity, which is essential for proper thyroglobulin function, rather than from direct apical iodide transport.
We also speculate that under high TSHR stimulation, SLC26A7 helps to maintain electrochemical gradients, intracellular pH, and fluid homeostasis in thyrocytes. Further studies are needed to clarify its protein interactions, regulatory pathways, and pH-modulatory effects.
In conclusion, we describe variable phenotypes associated with SLC26A7 variants, ranging from severe CH with large congenital goiters to delayed-onset hypothyroidism with dentofacial abnormalities. The thyrotropin-, GNAS-, and iodine-dependent regulation of SLC26A7 expression may contribute to this phenotypic variability.
Authors’ Contributions
L.N.: Writing—original draft (lead); patient recruitment—(equal); phenotyping and analyzation of clinical data (lead); and reviewing the final draft—equal. J.O.: Histological analyses (lead); writing—original draft (equal); and reviewing the final draft—equal. R.R.: Genetic analyses (lead); writing—original draft (equal); and reviewing the final draft (equal). V.M.: Phenotyping and analyzation of clinical data (equal) and reviewing the final draft (equal); V.L.: Analyzation of RNA sequence data (lead) and reviewing the final draft (equal); S.P.: Phenotyping and analyzation of clinical data (equal) and reviewing the final draft (equal); M.J.: Phenotyping and analyzation of clinical data (equal); patient recruitment (equal); histological analyses (equal); and reviewing the final draft (equal); S.T.: Histological analyses (equal) and reviewing the final draft (equal); C.L.: Genetic analyses (equal) and reviewing the final draft (equal); K.P.: Genetic analyses (equal) and reviewing the final draft (equal); K.M.: Patient recruitment—(equal); phenotyping and analyzation of clinical data (equal); and reviewing the final draft (equal); J.J.: Patient recruitment—(equal) and phenotyping and analyzation of clinical data (equal) and reviewing the final draft (equal); E.D.: Patient recruitment—(equal); phenotyping and analyzation of clinical data (equal); and reviewing the final draft (equal); H.H.: Patient recruitment—(equal); phenotyping and analyzation of clinical data (equal); and reviewing the final draft (equal); H.N.: Patient recruitment—(equal); phenotyping and analyzation of clinical data (equal); and reviewing the final draft (equal); L.V.: Patient recruitment—(equal); phenotyping and analyzation of clinical data (equal); and reviewing the final draft (equal); A.K.: Patient recruitment—(equal); phenotyping and analyzation of clinical data (equal); and reviewing the final draft (equal); P.M.: Patient recruitment—(equal); phenotyping and analyzation of clinical data (equal); and reviewing the final draft (equal); N.S.: Histological analyses (equal) and reviewing the final draft (equal); M.P.R.: Analyzation of FinnGen data and reviewing the final draft (equal); J.K.: Conceptualization (lead); writing—original draft (lead); patient recruitment—(equal); phenotyping and analyzation of clinical data (equal); genetic analyses and interpretation (equal); and reviewing the final draft (equal).
Footnotes
Acknowledgments
The authors thank the organizations supporting the study with grants from the Finnish Pediatric Foundation (no. 190001), grant from Turku University Hospital (no. 13527), Sigrid Juselius Foundation (12/2022), Jalmari and Rauha Ahokas Foundation, Novo Nordisk Foundation (no. 0078329) (J.K.), Doctoral Programme in Clinical Research (DPCR) (K.M.), and Drug Research Doctoral Programme (DRDP) (V.M.) of University of Turku. The authors thank Meri Davidsson for technical assistance with the histology specimens. The authors thank the personnel of the Institute of Biomedicine and Histology Core Facility of the Institute of Biomedicine, University of Turku, Finland for expertise and technical assistance in various stages of this study.
The authors want to acknowledge the participants and investigators of the FinnGen study. Following biobanks are acknowledged for delivering biobank samples to FinnGen: Auria Biobank (www.auria.fi/biopankki), THL Biobank (www.thl.fi/biobank), Helsinki Biobank (www.helsinginbiopankki.fi), Biobank Borealis of Northern Finland (https://www.ppshp.fi/Tutkimus-ja-opetus/Biopankki/Pages/Biobank-Borealis-briefly-in-English.aspx), Finnish Clinical Biobank Tampere (www.tays.fi/en-US/Research_and_development/Finnish_Clinical_Biobank_Tampere), Biobank of Eastern Finland (www.ita-suomenbiopankki.fi/en), Central Finland Biobank (www.ksshp.fi/fi-FI/Potilaalle/Biopankki), Finnish Red Cross Blood Service Biobank (www.veripalvelu.fi/verenluovutus/biopankkitoiminta), Terveystalo Biobank (www.terveystalo.com/fi/Yritystietoa/Terveystalo-Biopankki/Biopankki/), and Arctic Biobank (https://www.oulu.fi/en/university/faculties-and-units/faculty-medicine/northern-finland-birth-cohorts-and-arctic-biobank). All Finnish Biobanks are members of BBMRI.fi infrastructure (https://www.bbmri-eric.eu/national-nodes/finland/). Finnish Biobank Cooperative-FINBB (https://finbb.fi/) is the coordinator of BBMRI-ERIC operations in Finland. The Finnish biobank data can be accessed through the Fingenious® services (![]() ) managed by FINBB.
) managed by FINBB.
Author Disclosure Statement
L.N.: No conflict of interest; J.O.: No conflict of interest; R.R.: No conflict of interest; V.M.: No conflict of interest; V.L.: No conflict of interest; S.P.: No conflict of interest; M.J.: No conflict of interest; S.T.: No conflict of interest; C.L.: No conflict of interest; K.P.: No conflict of interest; K.M.: No conflict of interest; J.J.: No conflict of interest; E.D.: No conflict of interest; H.H.: No conflict of interest; H.N.: No conflict of interest; L.V.: No conflict of interest; A.K.: No conflict of interest; P.M.: No conflict of interest; N.S.: No conflict of interest; F.F.: No conflict of interest; M.P.R.: No conflict of interest; and J.K.: No conflict of interest.
Funding Information
This study was supported by grants from the Finnish Pediatric Foundation (J.K. no. 190001), grant from Turku University Hospital (J.K. no. 13527), Sigrid Juselius Foundation (J.K. 12/2022), Jalmari and Rauha Ahokas Foundation, Novo Nordisk Foundation (J.K. no. 0078329), Doctoral Programme in Clinical Research (DPCR) (K.M.), and Drug Research Doctoral Programme (DRDP) (V.M.) of University of Turku. N.S. is funded by the Wellcome Trust (Senior Fellowship 219496/Z/19/Z) and NIHR Cambridge Biomedical Research Centre. The FinnGen project is funded by two grants from Business Finland (HUS 4685/31/2016 and UH 4386/31/2016) and the following industry partners: AbbVie Inc., AstraZeneca UK Ltd, Biogen MA Inc., Bristol Myers Squibb (and Celgene Corporation & Celgene International II Sàrl), Genentech Inc., Merck Sharp & Dohme LCC, Pfizer Inc., GlaxoSmithKline Intellectual Property Development Ltd., Sanofi US Services Inc., Maze Therapeutics Inc., Janssen Biotech Inc, Novartis AG, and Boehringer Ingelheim International GmbH.
There is no funding information to declare for this study for L.N., J.O., V.L., S.P., M.J., S.T., C.L., K.P., J.J., E.D., H.H., H.N., L.V., A.K., P.M., F.F., and M.P.R.
Supplemental Material
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.