
Abstract
Mechanochromic polymers respond to mechanical forces with a change in color or luminescence and are potentially useful for applications that range from soft robotics to damage-sensing. Mechanochromic responses also permit the investigation of defect origination and propagation and other mechanical phenomena. In the last two decades, the incorporation of so-called mechanophores—molecules that undergo a specific chemical or physical change in response to mechanical force—has emerged as a widely employed design approach to render polymers mechanochromic. While the activation of most mechanophores is based on the cleavage of covalent bonds, supramolecular mechanophores leverage non-covalent interactions and change their molecular conformation or the interactions between multiple optically active moieties. This mini-review provides a historical perspective on my group’s 25-year-long journey in developing such motifs and integrating them into polymers.
This paper is dedicated to Byung-Lip (Les) Lee on the occasion of his retirement.
1. Introduction
The possibility of revealing mechanically induced events in polymers through optical changes enables fundamental investigations of mechanical phenomena, including defect formation and growth (Chen Y et al., 2021). Mechanochromic effects also hold significant potential for technological applications, including autonomous damage detection and soft robotics (Guo and Zhang, 2021). The earliest example of a mechanochromic material dates back a century when photoelasticity (stress-induced birefringence) in celluloid was discovered (Coker and Chakko, 1921). The effect facilitates the dynamic visualization of mechanically induced orientation in transparent polymers and has become widely employed for the analysis of stress distributions and fracture mechanisms (Ramesh, 2021). Microcapsules filled with latent dyes that are released and activated when the capsules are damaged represent another early approach to access mechanochromic materials. This framework, which has been exploited in carbonless copy paper and pressure-sensitive recording sheets for a long time (Green and Schleicher, 1957) was repurposed by White and co-workers to bestow polymers with healability and mechanochromic behavior (White et al., 2001) and the approach became more widely used (Calvino and Weder, 2018). Mechanochromic effects have also long been known to occur in deformable structurally colored materials (Kimura et al., 1979), for example, multilayered polymer films and fibers, and are the result of mechanically induced changes in the lattice distance of the photonic structures (Clough et al., 2021).The first molecular approaches to mechanochromic polymers were reported by Rubner (Nallicheri and Rubner, 1991; Rubner, 1986) and Reneker (Kim and Reneker, 1991) some 40 years ago, who demonstrated mechanochromic effects in thermoplastic polyurethanes containing covalently incorporated, optically active moieties. While the color changes displayed by the poly(diacetylene) segment-containing polymers investigated by Rubner were related to morphological changes, the approach pursued by Reneker involved the mechanically triggered conversion of cis-azobenzenes into trans isomers.
2. Mechanochromism based on excimer-forming dyes
My group’s interest in mechanochromic polymers was sparked in 2001 when we serendipitously discovered that fluorescent oligo(phenylene vinylene) dyes carrying electron-withdrawing cyano-groups on the vinylene bridges (Figure 1, cyano-OPVs) have a strong propensity to form excimers so that their solutions and aggregates or crystals exhibit very different emission spectra (Löwe and Weder, 2002a). We had just demonstrated that the deformation of blends of polyethylene and a conjugated polymer can cause the dispersion of aggregates of the latter (Trabesinger et al., 2000) and—initially unaware of the works cited above—surmised that the combination of the two concepts might allow access to mechanochromic luminescent polymers. Indeed, blends of small amounts of such aggregachromic dyes and low-density poly(ethylene) display the characteristic red or yellow excimer fluorescence associated with the aggregated state of the two different cyano-OPVs that were used, and deformation past the yield point (ca. 100% strain), triggered a striking change to green or blue emission, due to the dispersion of the dye molecules (Löwe and Weder, 2002b). We discovered quickly that controlling the nucleation process is important for the formation of small aggregates that efficiently disperse upon deformation of the host polymer and developed processes and tailored dyes to optimize the performance of such blends (Figure 1; Crenshaw and Weder, 2003; Kinami et al., 2006). We also realized early on that the ratio of the monomer and excimer emission intensities can be used as a ratiometric signal that is largely independent of the measurement parameters and makes the quantitative assessment of the mechanophore activation relatively straightforward (Crenshaw and Weder, 2003). Many of the supramolecular mechanophores that we subsequently developed mirror this feature. In the case of polyethylene blends with cyano-OPVs, the emission spectra show a clearly discernible reduction of the relative excimer emission intensity at strains as low as 10%, but the most pronounced spectral changes occur around the yield point (ca. 100% strain).
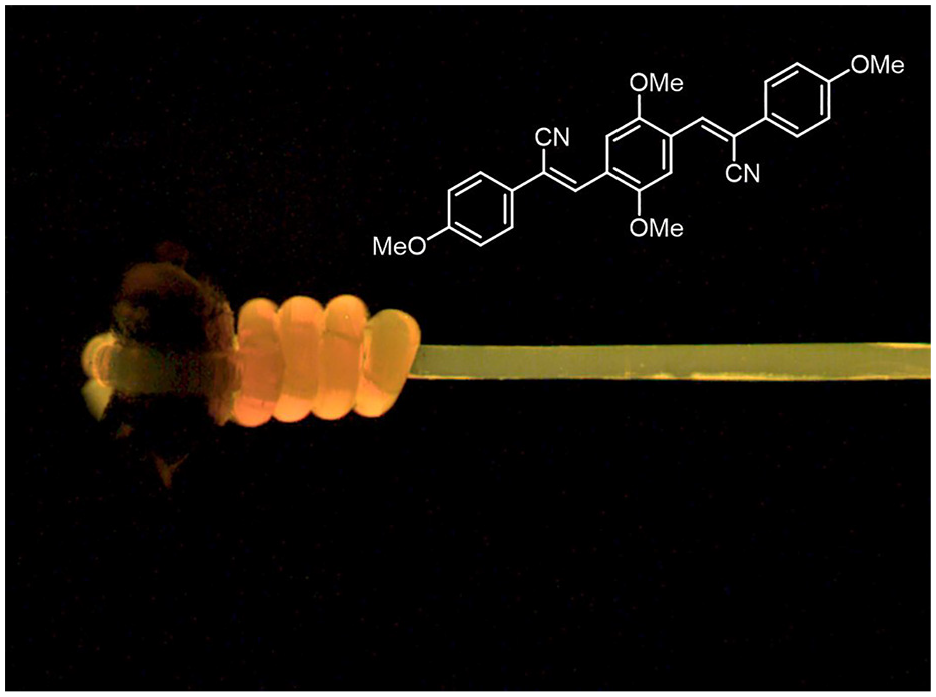
Picture of a mechanochromic fiber based on polyethylene and the cyano-OPV dye shown. Green emission reveals deformation.
While the concept of excimer-forming mechanochromic additives was successfully applied by us and eventually others to a range of polymer hosts and fluorescent dyes (Ciardelli et al., 2013; Haehnel et al., 2015; Sagara et al., 2016), it is not a priori generalizable, as the formation of the dye aggregates and their mechanically induced dispersion depend strongly on the nature of the polymer host. To address this limitation, we covalently incorporated cyano-OPVs into the backbone of different polymers (Crenshaw and Weder, 2006; Kunzelman et al., 2009), based on the expectation that such integration would couple the mechanoresponse more intimately to the polymer matrix. Indeed, pronounced mechanochromic effects were demonstrated in poly(ethylene terephthalate) and an elastic polyurethane containing covalently incorporated cyano-OPVs in their backbone. The mechanochromic response of the latter material mirrored its stress−strain behavior and was largely reversible, while the corresponding reference blends only displayed a modest fluorescence color change.
3. Supramolecular mechanophores
In the mid-2000s, parallel to our above-summarized efforts on mechanochromic blends based on excimer-forming dyes, the groups of Sijbesma (Paulusse and Sijbesma, 2004), Craig (Yount et al., 2005), and Moore (Berkowski et al., 2015) pioneered the incorporation of mechanically responsive motifs that would eventually become known as mechanophores (De Bo, 2020) into polymers and investigated how their mechanically induced scission could be controlled and applied in a constructive manner (Li et al., 2015). Davis et al. (2009) reported that this mechanism can be used to create mechanochromic polymers. In a landmark study, the team incorporated a spiropyran equipped with strategically placed reactive groups into the backbone of rubbery poly(methyl acrylate) and glassy poly(methyl methacrylate) and demonstrated that the reversible ring-opening reaction from a colorless spiro- into a colored merocyanine form is achievable through mechanical activation. Motivated by this work, countless researchers embarked on the development of mechanochromic mechanophores that undergo similar ring-opening reactions, as well as scissile mechanophores in which bonds are irreversibly cleaved (Huang et al., 2023). These motifs are either covalently introduced into the polymer backbone or incorporated as cross-links and are activated if the applied force or strain exceeds a threshold. However, the activation of many of these mechanophores is not, or not fully reversible, as their activation mechanism involves the cleavage of covalent bonds, whose restoration may be kinetically or thermodynamically stifled. Moreover, many mechanophores that undergo a chemical change can also be activated by other forms of energy, notably UV light and heat. In other words, their response in a polymer is not necessarily specific to an applied force.
To address these limitations, we eventually embarked on the development of supramolecular mechanophores whose activation is based on physical changes (Kiebala et al., 2021). In a long-lasting collaboration with Yoshi Sagara, we took the mechanically induced spatial separation of excimer-forming dyes and other electronically interacting motifs—quencher-emitter pairs, as well as charge-transfer complexes—from an intermolecular (Crenshaw and Weder, 2006) to an intramolecular level. As a first embodiment, we reported in 2018 a mechanochromic rotaxane that features a dumbbell-shaped axle molecule containing naphthalenetetracarboxylic diimide (NPI) as a quencher and a 1,5-dinaphtho[38]crown-10 macrocycle equipped with a 4,7-bis(phenylethynyl)-2,1,3-benzothiadiazole (BTH) as fluorophore (Figure 2, Rotaxane 1; Sagara et al., 2018). Hydroxyl groups attached to the two parts of the mechanically interlocked molecule allowed its incorporation into an elastic polyurethane. In the force-free state, the BHT fluorophores reside near the NPI quenchers, and their emission is largely turned off. Deformation of the polymer causes the spatial separation of the fluorophore/quencher pairs in the mechanophores and thus leads to a fluorescence turn-on. Because this separation cannot be caused by exposure to light or heat, the mechanophore is exclusively activated by mechanical force. The process elicits an easily detectable optical signal that correlates well with the macroscopically applied force and is instantly reversible, that is, the emission is turned off once the force is removed and the elastic polymer relaxes. In spite of the (presumably) low activation energy of the rotaxane mechanophore, its activation in the polyurethane requires high strain levels. In the elastic polyurethane, even at a strain of 600% and an applied stress of ca. 20 MPa, only about a third of the mechanophores are activated. This is related to the entropically favored, coiled chain conformation that the macromolecules adopt and first must extend, before the mechanophores experience a force that is sufficiently high activate them.
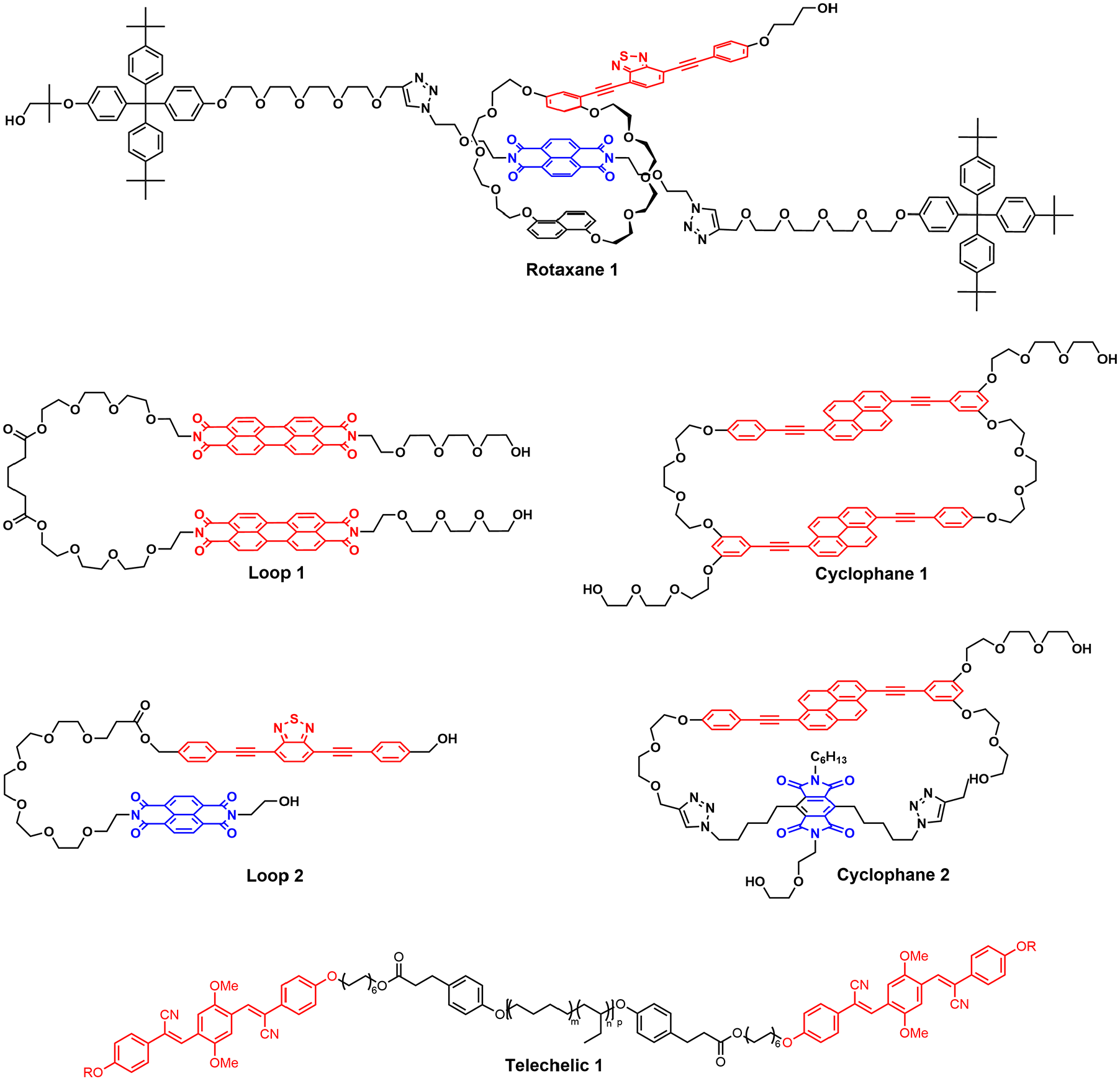
Chemical structures of the supramolecular mechanophores discussed.
By changing the emitter, we demonstrated another advantage of supramolecular mechanophores, that is, that their optical response can be modified in a rational manner (Sagara et al., 2019). Blue, green, and red-emitting mechanophores based on the initial rotaxane design were made by substituting the original emitter with a 1,6-disubstituted pyrene, a 9,10-disubstituted anthracene, and 4-(dicyanomethylene)-2-methyl-6-(4-dimethylaminostyryl)-4H-pyran. We blended three polyurethanes that each contain one of these mechanophores and demonstrated that the deformation of such blends leads to white emission. While the three motifs do not necessarily have the same activation energy, they exhibit identical responses to a macroscopically applied stress/strain when incorporated in the same polymer. By contrast, the mechanical (de)activation behavior of rotaxane mechanophores is affected by the length of the axle (Mori et al., 2023) and the size of the stoppers (Muramatsu et al., 2021). Polymers containing rotaxanes with longer axles show a slower reduction of the fluorescence intensity upon stress relaxation than their counterparts with shorter mechanophores, reflecting that the relaxation is subject to kinetic effects. Small stoppers allow for the irreversible de-threading of the cycles and enable a combination of reversible and irreversible effects.
The most significant drawback of rotaxane mechanophores is their exceedingly complicated synthesis, which requires more than 20 steps. We therefore developed several cyclophane mechanophores, which are easier (but also not easy) to access (Sagara et al., 2021; Thazhathethil et al., 2022). The first cyclic motif that we explored features two fluorescent 1,6-bis(phenylethynyl)pyrene moieties, which, in the stress-free state, form intramolecular excimers (Figure 2, Cyclophane 1). Films of a polyurethane containing this motif in the backbone display some excimer emission, although the extent of monomer emission is much higher than observed in solutions of this polymer. This difference is likely caused by the kinetic trapping of some of the mechanophores in conformations in which excimer formation is not feasible. Nevertheless, upon stretching the polymer, an instant and reversible change in the emission spectra is observed that is consistent with the mechanically induced separation of the excimer-forming emitters. However, the changes are not very pronounced and not easily discernible by the unassisted eye. To overcome this problem, we devised another cyclophane-based mechanophore comprised of a 1,6-bis(phenylethynyl)pyrene luminophore emitting blue light and a pyromellitic diimide that was originally chosen as a quencher (Figure 2, Cyclophane 2). Instead, we discovered the formation of a charge-transfer (CT) complex that emits red-orange light. Gratifyingly, the orange CT-complex emission remains dominant in films of a polyurethane containing the cyclophane, while deformation causes an easily detectable change of the emission color to a deep blue. Interestingly, the activation behavior of the two cyclophanes—when incorporated in the same polymer—is rather different. Overlays of the strain-stress curves and the optical signals show that in the case of Cyclophane 1, the optical signal correlates with the applied strain while the response of Cyclophane 2 is a better indicator of the applied stress. We related the differences in activation behavior to the fact that the charge-transfer interactions in Cyclophane 2 are stronger than the π-π interactions between the excimer-forming luminophores in Cyclophane 1, which may lead to a higher activation force. However, this was not experimentally verified. By contrast, we demonstrated that the ring size of the CT cyclophane mechanophores significantly affects their activation behavior, likely by dictating to what extent the two optically active motifs are able to interact in the idle state and upon applying a force and deforming the motif (Thazhathethil et al., 2024).
With the goal of creating supramolecular mechanophores that display similar features as the cyclophanes discussed above but are still easier to synthesize, we devised a loop-forming motif in which two fluorescent, excimer-forming perylene diimide (PDI) moieties are covalently connected by a short linker (Figure 2, Loop 1; Traeger et al., 2021). In the idle state, attractive interactions among the PDIs cause the motif to fold into a loop so that the emission is excimer-dominated. The loop-forming mechanophore was equipped with reactive groups and covalently incorporated into the backbone of poly(methyl acrylate) as well as different polyurethanes (Traeger et al., 2021, 2022a). Gratifyingly, the mechanophore performs well in all matrices, that is, the fraction of PDI monomer emission intensity increases upon deformation to the extent that the effect is clearly visually discernible. However, the specific mechanoresponse depends on the polymers’ properties. The most pronounced response is observed for a polyurethane with the highest strength and stiffness. By contrast, a similar motif that we expected to display a fluorescence turn-on, containing the NPI quencher and the BTH fluorophore that were used in Rotaxane 1 instead of the excimer-forming PDIs employed in Loop 1, did not perform as desired (Figure 2, Loop 2), bringing out an important design criterion that we are currently addressing (Traeger et al., 2022b). Clearly, the NPI-BTH interactions are not sufficiently strong to drive the formation of loops, so that, similar to Cyclophane 1, “activated” conformations are trapped during polymer processing.
4. Mechanochromic blends revisited
Driven by the appeal of mechanochromic additives that can be used in any given polymer without the need for covalent incorporation (Ciardelli et al., 2013; Haehnel et al., 2015; Sagara et al., 2016) we recently developed telechelic macromolecules that are end-functionalized with excimer-forming dyes, including the cyano-OPVs that got us started (Figure 2, Telechelic 1; Calvino et al., 2019; Oggioni et al., 2024). Originally, we speculated that these high-molecular-weight probes would mix and entangle with the matrix polymer, that nucleation-dominated, transport-limited aggregation processes would cause their luminescent termini to form small aggregates, and that the anticipated entanglements with the matrix would promote efficient stress transfer and disassembly of the latter. Indeed, we were able to show that blends of small amounts of the additives and different host polymers, including poly(ε-caprolactone), polyisoprene, poly(styrene-b-butadiene-b-styrene), and different thermoplastic polyurethanes, display highly sensitive, reversible, strain-dependent mechanochromic responses. However, the underlying mechanism is wildly different than initially thought (Kiebala et al., 2023). Confocal microscopy revealed that the additives microphase-separate from the matrices and form discrete inclusions with diameters of a few hundred nanometers. The mechanochromism of the blends originates from the distortion of these inclusions, which, independent of the content of the telechelic additive and the nature of the matrix, deform homogeneously and reversibly in response to a macroscopically applied strain. Notably, this approach has allowed the detection of strains as low as 5%. While it remains to be determined why the inclusions change their emission characteristics upon deformation and to what extent their optical response relaxes, it appears that such universal additives are uniquely suited for the straightforward fabrication of mechanochromic blends.
5. Conclusions, open questions, and unsolved problems
While the first successful molecular concepts to create mechanochromic polymers were all rooted in non-bond-breaking approaches, fluorescence- and/or color-changing mechanophores that operate on the basis of covalent bond cleavage have become the most widely employed framework approach to render polymers mechanochromic. Supramolecular mechanophores, in which interactions between multiple optically active moieties and, thereby, the optical characteristics of this ensemble, are controlled by an applied force, combine elements of both designs. Many of the embodiments presented here exhibit responses that are instantaneous, fully reversible, and exclusively triggered by mechanical stress or strain. The design approach of these motifs is modular, that is, elements such as the optically active moieties or the size of the axle, cycle, or loop can be modified to rationally control the optical response to mechanical activation.
While elastic polyurethanes and poly(methyl acrylate) were used as testbeds in most of our studies, mainly with the goal of exploring to what extent the activation of the mechanophore is reversible and how well the nonlinear stress-strain curves can be mirrored by mechanophore activation, the supramolecular mechanophores reported here should also function in other matrices, but this has yet to be demonstrated. In this context, it will be especially interesting to explore how well these motifs are suited to probe irreversible mechanical events and if their ability to relax poses a problem. Notwithstanding minor optical changes that result from ground-state interactions, all mechanophores reported here comprise fluorescent dyes and change their fluorescence color or intensity upon activation. Similar supramolecular mechanophores, which change their absorption characteristics and thus cause an easily detectable color change, have to our best knowledge yet to be developed.
Interestingly, after more than 20 years of intense international research, the mechanistic and quantitative understanding of mechanically induced processes in mechanophore-containing polymers remains limited and technological applications are, to our best knowledge, still absent. Even (seemingly) simple products, such as damage-indicating fibers (Figure 1) are waiting to be realized, which in turn reflects that the field still offers ample problems that are waiting to be solved. Based on our experience, they include the need for a better understanding of stress-strain-activation relationships, better/simple mechanophores, increasing the fraction of mechanophores that are activated, the ability to achieve mechanophore activation at low strain, and simple(r) ways to incorporate mechanophores into industrial polymers that may experience harsh processing conditions without being prematurely activated.
Footnotes
Acknowledgements
I am indebted to the many students, postdocs, and collaborators who made this work possible and appear in the referenced publications. I also would like to thank Iulia Scarlat for help with the figures. Our recent work on mechanochromic polymers has been supported through the National Center of Competence in Research (NCCR) Bio-Inspired Materials, a research instrument of the Swiss National Science Foundation (SNSF) under Grant No. 51NF40_182881, the Army Research Office under Grant No. W911NF-23-2-0075, the Swiss State Secretariat for Education, Research and Innovation (SERI) through the Bilateral Science and Technology Cooperation Program with Asia (Grant No. EG_JP_special_032023_22), and the Adolphe Merkle Foundation. Support from the US Air Force Office of Scientific Research under Grant No. FA9550-23-1-0250 is also acknowledged.
Declaration of conflicting interests
The author declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author received no financial support for the research, authorship, and/or publication of this article.
Data availability statement
Data sharing not applicable to this article as no datasets were generated or analyzed during the current study.