
Abstract
Peste-des-petits-ruminants (PPR) is primarily a disease of small ruminants caused by peste-des-petits-ruminants virus (PPRV; Paramyxoviridae, Morbillivirus caprinae), formerly the small ruminant morbillivirus. PPRV can cause significant morbidity and mortality in small ruminants and a significant economic impact. Conventional reverse-transcription PCR (RT-PCR), and probe-based and SYBR Green–based RT quantitative real-time PCR (RT-qPCR), are employed for the molecular detection of PPRV. Here we describe a SYBR Green–based RT-qPCR for rapid and sensitive detection of PPRV. We designed the specific primers from the conserved region of the fusion gene (F) of PPRV. The standard curve of the established RT-qPCR assay had a good linear relationship. The developed assay was also 3 log units more sensitive than the conventional RT-PCR, with a detection limit of 13.6 copies and an efficiency of 98.2%. There was no cross-reactivity with other caprine respiratory viruses, namely bluetongue virus, goatpox virus, and orf virus. The positive detection rate of clinical samples was 11 of 64 (17.2%) versus 10 of 64 (15.6%) by conventional RT-PCR. We confirmed our results by sequencing the full F and N genes of the isolates. Our SYBR Green RT-qPCR can be used as a fast, economical, and sensitive alternative to RT-PCR for the detection of PPRV.
Peste-des-petits-ruminants (PPR) is a WOAH-listed disease of small ruminants that poses a significant threat to the world’s small ruminant population. Disease outbreaks can be detrimental to goat production, which could create an economic crisis worldwide, particularly in developing countries, and the disease requires more attention wherever small ruminants play a major role in the agricultural sector.11,15 The disease has a very high morbidity of up to 100% and mortality of up to 90%. 17 The affected animals have pyrexia, oculonasal discharge, pneumonia, and diarrhea as initial clinical signs, and ulceration of the mucous membranes, and gastroenteritis on autopsy. Following the first report of PPR from Côte d’Ivoire in 1942, the disease has been reported in Africa, the Middle East, Europe, and Asia. 8
The genome of the peste-des-petits-ruminants virus (PPRV; Paramyxoviridae, Morbillivirus caprinae) consists of 6 transcription units that code for the nucleocapsid protein (NP), phosphoprotein (P), matrix protein (M), fusion protein (FP), hemagglutinin protein (HP), and large protein (L). The large protein along with the P protein forms the viral RNA-dependent RNA polymerase.3,4 Among the 4 lineages, lineages I and II were reported mostly from western and central Africa, lineage III from the Arabian Peninsula, eastern Africa, and southern India, and lineage IV mostly from Asia, extending from the Middle East to Tibet. 16
Various reverse-transcription PCR (RT-PCR) assays have been used for the detection of PPRV, but these methods are being replaced by more sensitive and specific RT quantitative real-time PCR (RT-qPCR) assays targeting N, F, and M genes.5,9,13 The main advantages of RT-qPCR over conventional RT-PCR are that it is less labor intensive, does not require gel documentation, and has a low chance of contamination, which has led to its widespread acceptance. 6 A few RT-qPCR assays using SYBR Green chemistry have been described for detecting and quantifying PPRV in clinical samples targeting the M and N genes.1,2,5
Although the F gene is one of the most antigenically conserved of the morbillivirus proteins, the nucleotide sequence is not highly conserved between viruses because of the redundancy of the genetic code. This enables the selection of primer sequences that are specific to each virus. 9 We established a highly sensitive SYBR Green RT-qPCR assay targeting the conserved homologous sequence of the F gene for the detection of PPRV, and evaluated its specificity, reproducibility, and applicability for clinical testing.
Materials and methods
Primer design
Sequences of PPRV lineages I–IV from different countries available in GenBank were aligned using MEGA11 software. 10 We designed primers for our SYBR Green RT-qPCR from the conserved F gene region of the aligned sequences using OligoAnalyser (IDT, https://www.idtdna.com/pages/tools/oligoanalyzer) as follows: PPR FP500FPKD (5′-ACCATACTGGCAGTACAGG-3′; covering nucleotides 499–517) and PPR FP589RPKD (5′-ACCTACCARCTCRCATGAC-3′; complementary to nucleotides 567–585). The expected amplicon size of the product was 87 bp. The oligos were synthesized by MilliporeSigma. For amplification of the complete F gene, a forward primer (PPR FPFKD: 5′-ACATCCATGCGTAAACATYATGAC-3′) with 24 nucleotides was designed from conserved sequences that included 18 nucleotides from intergenic sequences and the first 6 nucleotides of the F gene. A reverse primer (PPR FPRKD: 5′-CTACAGTGATCTYACGTAMGACTTTGAG-3′) with 28 nucleotides was designed from position 1614–1641 of the F gene.
Sample collection
Nasal and conjunctival swabs and blood were collected from live animals brought to the Teaching Veterinary Clinical Complex (Kerala Veterinary and Animal Sciences University [KVASU], Pookode, India) with respiratory difficulty, and from field outbreaks in Kerala, India. Our study was performed per the guidelines for the care and use of animals by the Institutional Animal Ethics Committee of the College of Veterinary and Animal Sciences, KVASU, and the Animal Welfare Board of India. Tissue samples were collected from goats brought to the Department of Veterinary Pathology, College of Veterinary and Animal Sciences (KVASU) for autopsy. Samples of lungs, liver, spleen, and mesenteric and mediastinal lymph nodes were collected in viral transport medium and stored at −80°C until processing. We collected 64 samples from suspect animals.
Extraction of total RNA from clinical samples
Total RNA was isolated from the tissue samples, nasal and conjunctival swabs, and blood (GeneJET RNA purification kit; Thermo Scientific) per the manufacturer’s protocol and stored at −20°C until further processing.
Synthesis of complementary DNA by reverse transcription of total RNA
Complementary DNA (cDNA) was synthesized (RevertAid first strand cDNA synthesis kit; Thermo Scientific) per the manufacturer’s protocol. We added the following to a PCR tube kept on ice: 2 µL of RNA, 1 µL (0.2 µg/µL) of random hexamer primer, and nuclease-free water (NFW) to make the volume up to 12 µL. The mixture was incubated at 65°C for 5 min. The above mixture was cooled to 25°C and 4 µL of 5× reaction buffer, 1 µL (20 U/µL) of Ribolock RNase inhibitor, 2 µL of 10 mM dNTP mix, and 1 µL (200 U/µL) of RevertAid H minus M-MuLV reverse transcriptase were added; then the mixture was incubated at 25°C for 5 min followed by 60 min at 42°C. The reaction was terminated by heating at 70°C for 5 min. The cDNA was stored at −20°C for further use.
Preparation of standard plasmid DNA templates
Amplification of 1,660 bp of the F gene was carried out using RT-PCR. The RT-PCR was carried out in a reaction volume of 25 μL containing 5 μL of 5× SuperFi buffer, 1.25 μL of forward primer, 1.75 μL of reverse primer (10 pmol each), 2 μL of cDNA (500 ng), 0.5 μL of dNTP (10 mM), 0.25 μL of Platinum SuperFi DNA polymerase, and NFW. The RT-PCR products were purified using a PCR gel extraction kit. The amplicon was cloned using a CloneJET PCR cloning kit per the manufacturer’s guidelines and transformed into Escherichia coli DH5α competent cells. From the recombinant E. coli, the plasmid DNA was isolated using the Miniprep kit.
Calculation of copy number
The plasmid DNA concentration was used to calculate the copy number of plasmid DNA using the following formula:
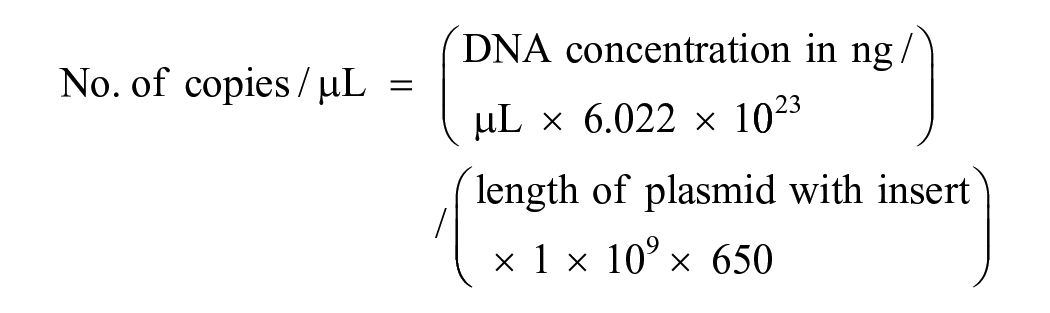
Standardization of SYBR Green RT-qPCR for detection of PPRV
Conventional gradient PCR was conducted using RT-qPCR primers with an annealing temperature range of 52–62°C. The optimum concentration of primers was checked using primer concentrations of 1, 2, 5, and 10 pmol. The reaction mixture was comprised of 12.5 µL of 2x EmeraldAmp GT PCR master mix (TaKaRa), 1 µL each of primers (10 pmol), 1 µL of plasmid DNA, and NFW to make the volume up to 25 µL. The cycling conditions were 95°C for 2 min (initial denaturation), 35 cycles of 94°C for 1 min (denaturation), gradient from 52–62°C for 30 s (annealing), and 72°C for 30 s (polymerization), followed by a single cycle at 72°C for 5 min (final extension). All of the PCR products were electrophoresed in 2% agarose gels with ethidium bromide and visualized using a gel documentation system. The annealing temperature that gave the thickest band without primer-dimer formation was selected for the RT-qPCR.
For RT-qPCR, the reaction mix contained 10 µL of Power Up SYBR Green real-time PCR master mix (Applied Biosystems), 1 µL each of 2 pmol forward and reverse primers, 1 µL of template plasmid DNA, and volume made up to 20 µL with NFW. The samples were run in a real-time PCR thermocycler (StepOnePlus; Applied Biosystems) programmed to record fluorescence. The cycling conditions for the RT-qPCR are as follows: 50°C for 2 min (Uracil-DNA glycosylases activation), 95°C for 2 min (initial denaturation), 40 cycles of amplification (95°C for 15 s for denaturation, 61°C for 1 min for annealing and extension), followed by melt curve analysis from 60°C to 95°C with 0.3°C increment.
Analytic sensitivity and specificity of RT-qPCR
Serial 10-fold dilutions (100–10−9) of the purified plasmid were made using NFW and used to determine the analytic sensitivity of our RT-qPCR based on copy number. RT-qPCR was performed with a volume of 20 µL, containing 10 µL of 2× Power SYBR mixture and 1 µL of plasmid in a 10-fold dilution series of 1.3 to 1.3 × 108 copies/µL. The final primer concentration was 2 pmol. Each program was composed of an initial step at 95°C for 10 min and 40 cycles of 95°C for 15 s, and 61°C for 1 min. Fluorescence was measured by a program that had been set in advance. The melting curves were generated by monitoring the fluorescence of the SYBR Green signal from 60–95°C. The highest dilution of plasmid DNA that could be detected in the RT-qPCR was determined from the Ct values obtained.
To check the reproducibility of the Ct values in the standardized RT-qPCR assay, standard plasmid DNA dilutions of 103–105 copies/µL were assayed in triplicate by RT-qPCR for the calculation of intra-assay CVs. The inter-assay was performed by 3 independent assays every other week using standard plasmid DNA dilutions of 104–106 copies/µL.
Detection of PPRV in clinical samples by RT-qPCR
For the detection of PPRV in clinical samples, RT-qPCR was performed with the standardized protocol using as template 1 µL of cDNA prepared from the 500 ng of RNA extracted from the clinical specimen. The SYBR Green RT-qPCR products were visualized by gel electrophoresis on 2% agarose gel to compare the sensitivity with the conventional PCR product. The representative 3 positive SYBR Green RT-qPCR products were cloned in a pJETl.2/blunt vector, and recombinant plasmids were sequenced to check the specificity of the amplicons. DNA or cDNA prepared from the RNA extracted from other respiratory viruses of goats [bluetongue virus (BTV), orf virus (ORFV), and goatpox virus (GPV)] were subjected to SYBR Green RT-qPCR to confirm the analytic specificity of our technique. RNA-free water was used as the negative control.
Statistical analysis
We compared the clinical sensitivity, specificity, and accuracy of our SYBR Green RT-qPCR with the F gene–based PCR using the following formulas:
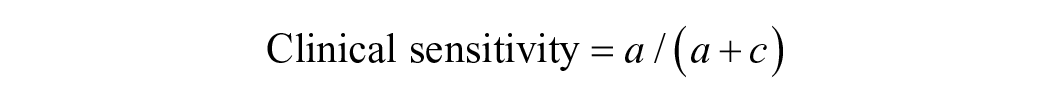

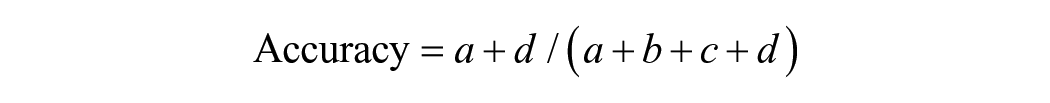
where a = the number of samples positive by both (i.e., the test to be compared and the gold standard test), b = the number of samples positive by the standard test and negative by the test to be compared, c = the number of samples negative by the standard test and positive by the test to be compared, and d = the number of samples negative by both.
The results obtained from the test were analyzed for the percentage of agreement with the F gene–based conventional PCR with the use of kappa statistics using SPSS v.24.0 (IBM). Kappa statistics are a decimal measure of agreement between 2 tests, especially in the absence of a standard, and are defined as kappa or κ.
Results
Generation of plasmid DNA templates
The 1,641-bp fragment of the F gene was amplified by RT-PCR with designed primers and cloned into a pJETl.2/blunt vector. The fragment was sequenced and the identity was confirmed by BLAST analysis. The concentration of recombinant plasmid was 6.94 ng/µL, and the copy numbers were 1.36 × 109 copies/µL.
Standardization of SYBR Green RT-qPCR
After the optimization of our SYBR Green RT-qPCR, a final concentration of 2 pmol of each primer had the highest efficiency and sensitivity. The SYBR Green RT-qPCR conditions for the detection of PPRV were standardized as follows: the reaction mix contained 10 µL of SYBR Green real-time PCR master mix (Applied Biosystems), 1 µL each of 2 pmol forward and reverse primers, 1 µL of template cDNA, and the volume was made up to 20 µL with NFW. The optimized annealing temperature was 61°C.
Standard curve analysis
A linear relationship between plasmid DNA and Ct values over a range of 101–108 copies was detected by our SYBR Green RT-qPCR. The efficiency of our assay was 98.2%, and the correlation coefficient (R2) of the standard curve was 1, with a slope of −3.365 (Fig. 1).
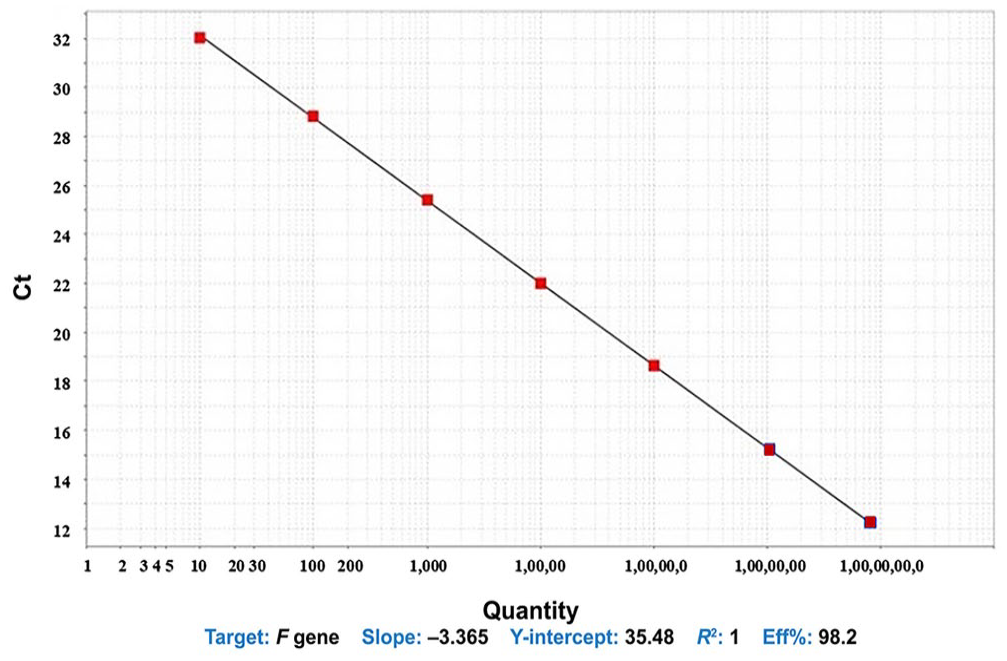
A linear relationship between Ct values and the dilutions of the plasmid DNA of the F gene of peste-des-petits-ruminants virus in the standard curve. The efficiency of our SYBR Green RT-qPCR assay was 98.2%, and the correlation coefficient (R2) of the standard curve was 1, with a slope value of the curve of −3.365.
Sensitivity, specificity, and reproducibility of RT-qPCR
The SYBR Green RT-qPCR products were visualized by gel electrophoresis. The limits of detection of our SYBR Green RT-qPCR and the conventional RT-PCR were 13.6 copies for which a Ct value of 33.8 was obtained (Fig. 2) and 1.35 × 103 copies (below this concentration no bands were visible in gel electrophoresis after RT-PCR), respectively. A Ct value of 35 was taken as a positive-negative threshold value for our SYBR RT-qPCR. Only PPRV was detected as a single melt peak at 76.5°C (Fig. 3), with no positive result for the 3 other caprine respiratory viruses (BTV, GPV, ORFV) or the negative control. The sequencing of 3 representative SYBR Green RT-qPCR–positive products in the pJETl.2/blunt vector confirmed the specificity of our RT-qPCR.
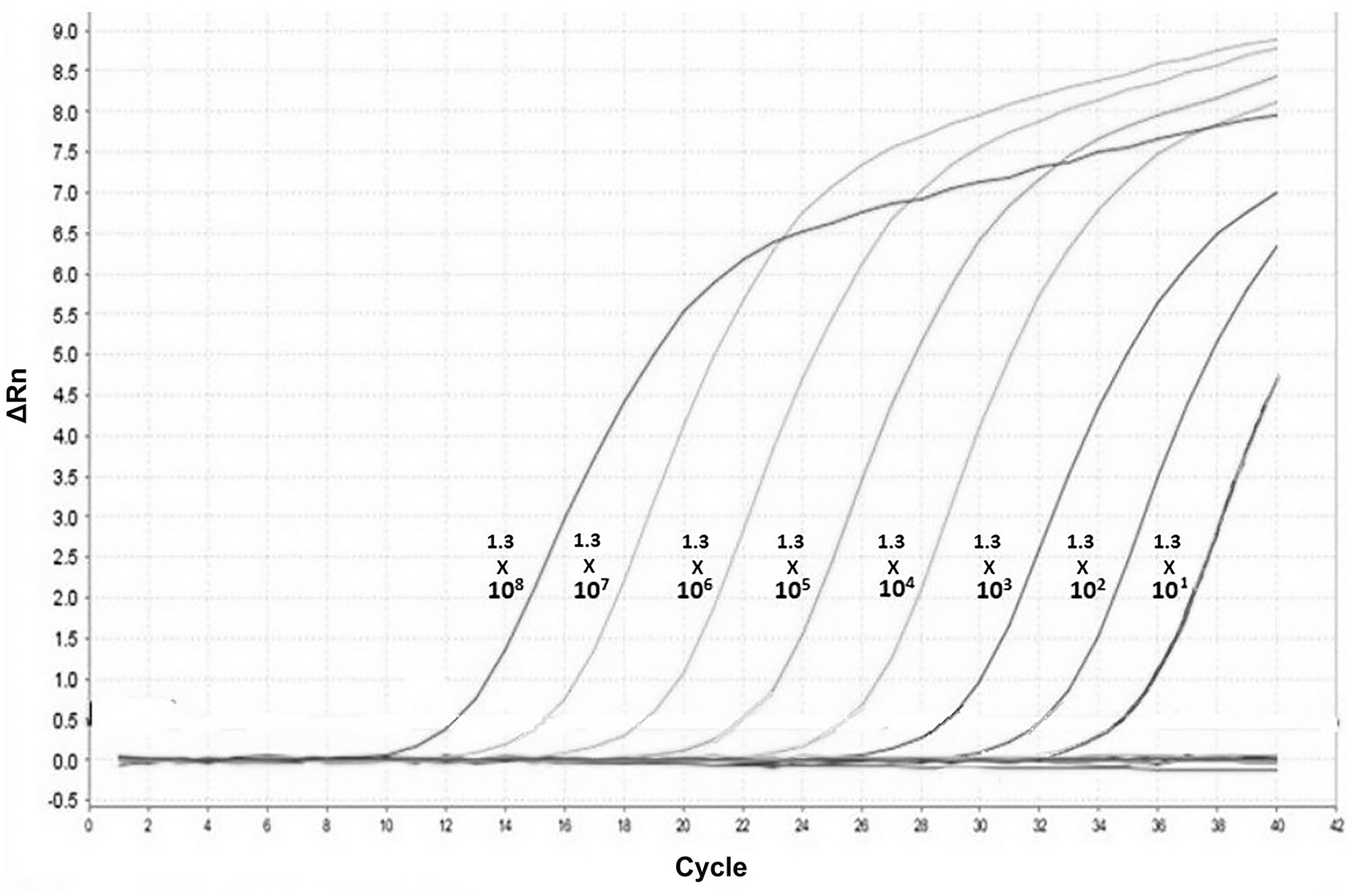
The amplification plot for different copy numbers of the plasmid. The minimum detection limit of our SYBR Green RT-qPCR was 13.6 copies for which a Ct value of 33.8 was obtained.
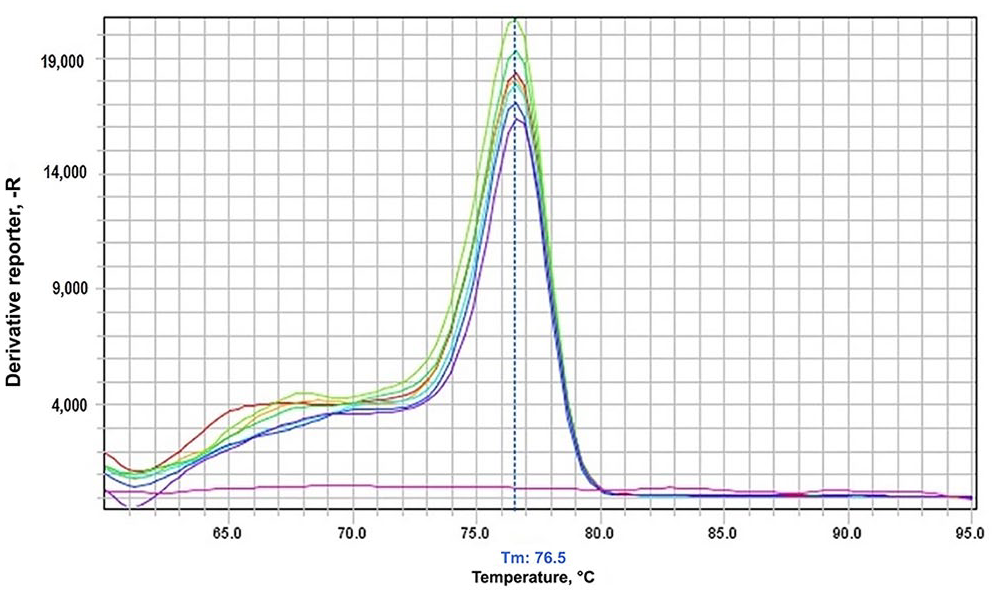
Only positive samples had a single peak at 76.5°C in the melt curve analysis; no other peaks for other viruses or the negative control.
The intra-assay CVs were 0.1–1.2% and the inter-assay CVs were 0.79–0.99% for standard dilutions of 104–106 copies/µL (Table 1).
Reproducibility of Ct values in intra- and inter-assays with 3 standard concentrations and CV%s in our RT-qPCR for peste-des-petits-ruminants virus.

For the intra-assay, 3 replicates of standards (103–105 copies/µL) were performed; the inter-assay was tested for each of 3 standards (104–106 copies/µL) every other week.
Detection of PPRV in clinical samples
Among the 64 samples that we tested with our SYBR Green RT-qPCR, 11 samples were positive and 53 were negative for PPRV. The Ct values of positive samples ranged from 26.6 ± 0.13 to 32.5 ± 0.22. In the conventional RT-PCR assay, only 10 samples were positive and 54 were negative. From the positive samples, the full N and F genes of the isolates were sequenced and submitted to GenBank (ON988192–ON988209, OR253783).
Statistical analysis of the RT-qPCR assay using kappa statistics
We estimated the sensitivity, specificity, and accuracy of our SYBR Green RT-qPCR to be 1, 0.98, and 0.98, respectively, compared with the F gene–based conventional RT-PCR. The κ value was estimated to be 0.92.
Discussion
We estimated the detection limit of our SYBR Green–based RT-qPCR assay targeting the F gene to be 13.6 copies, which is in line with the detection limit specified for several other assays (8.1 RNA copies per reaction, 6 10 genome copies per reaction 7 ) and is better than the detection limit of 100 copies by another RT-qPCR assay. 18 Our assay had sensitivity, specificity, and accuracy of 1, 0.98, and 0.98, respectively, compared with a conventional RT-PCR assay. Similar results were obtained for the RT-qPCR assays developed by other researchers.1,6 The estimated κ value was 0.92, which indicated perfect agreement. 12 The efficiency of our assay was 98.2%, which is well within the recommended range of 90–110%. We subjected our SYBR Green RT-qPCR products to gel electrophoresis, and no visible band was observed in the plasmid concentration from 1.36–1.36 × 103; hence, our assay has a 3 log greater sensitivity than the conventional RT-PCR. Our in silico analysis using primer-BLAST software showed that the designed primers can detect viruses from all 4 lineages of PPRV. However, we detected only lineage IV of PPRV, as other lineages are not prevalent in India.
Of the 64 samples tested by the SYBR Green RT-qPCR assay for the detection of PPRV targeting the F gene region, 11 samples were positive for PPRV. Percentage positivity was estimated to be 17.2%, which is greater than that obtained in conventional RT-PCR targeting the partial F gene. Our results are also in agreement with a previous study. 6
For the detection of PPRV nucleic acid from PPR-suspect clinical samples of sheep and goats, our SYBR Green assay was more sensitive, accurate, and rapid than a conventional RT-PCR. Our SYBR Green RT-qPCR could be used as an alternative to existing assays for PPRV and could be useful for rapid clinical detection.1,5 Also, our RT-qPCR could detect low viral loads during the early and late phases of a disease outbreak. 14
Given that SYBR Green I is comparatively cheaper than probe-based RT-qPCR, our assay could be used economically. Hence, our SYBR Green–based RT-qPCR based on the F gene can be employed for the economical, rapid, and sensitive detection of PPRV from field clinical samples, which in turn will help in implementing effective control measures to combat disease outbreaks.
Footnotes
Acknowledgements
We thank the Dean, College of Veterinary and Animal Sciences, Pookode, KVASU, for providing facilities and financial support for the conduct of the study.
Data availability
The datasets generated during and/or analyzed during our study are not publicly available but are available from the corresponding author upon reasonable request.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
Financial support for our study was provided by the College of Veterinary and Animal Sciences, Kerala Veterinary and Animal Science University, Pookode, Wayanad, Kerala.