
Abstract
The detection of Coxiella burnetii in ruminants remains challenging despite the use of new technology and the accumulation of novel knowledge. Serology tools, the primary methods of infection surveillance in veterinary medicine, have limitations. We used recombinant antigen production to develop an ELISA based on the SucB protein, one of the major immunodominant antigens described in humans and laboratory animals. We produced the antigen successfully in an Escherichia coli heterologous system, confirmed by sequencing and mass spectrometry, and seen as a band of ~50 kDa in SDS-PAGE and on western blot analysis. We compared the performance of the recombinant ELISA with a commercial ELISA. We observed agreement of 83.5% and a substantial Cohen κ value of 0.67 in our pilot study.
Q fever, an infectious disease caused by Coxiella burnetii, causes economic losses in ruminant herds and is a serious zoonosis. 1 C. burnetii infection is present on all 5 continents in a wide variety of animal species.8,18 The biggest challenges in eliminating this infection are subclinical infections in domestic animals (the main reservoirs are small ruminants) as well as a lack of coordinated surveillance and monitoring plans.2,25 Another issue with this disease is the diagnostic performance of serologic assays, mainly ELISA, given that complement fixation is now dated and immunofluorescence is not applied routinely on large numbers of animals for practical reasons. 34 ELISAs are often used as rapid screening tools to conduct epidemiologic and surveillance investigations in ruminants.19,27,30 Commercial ELISA kits are based on inactivated full antigen and are not able to distinguish between acute and chronic infections or between infected and vaccinated animals. Potential cross-reactivity and the antigenic variation of circulating Coxiella can affect, respectively, the specificity and sensitivity of routinely used assays.9,10,28 Moreover, in the early stages of infection, ruminants can shed the bacteria even if testing seronegative.27,30
Many attempts have been made to develop a recombinant ELISA to overcome performance limitations and to provide a tool that is simple to implement and does not require specific biosafety requirements.11,12,21,31 Given that these efforts have resulted in the development of tests with acceptable performance but not capable of outperforming commercial tests, it is necessary to continue researching new recombinant antigens because the eradication or control of Q fever requires the continuous improvement of tests. Dihydrolipoyllysine-residue succinyltransferase (SucB; ORF: CBU_1398; GenBank Q83BU7), is understudied although it has been identified as one of the key immunodominant antigens in humans and laboratory animals that is capable of identifying antibodies during the acute stage of disease.3,7,22 SucB, which is a component of the tricarboxylic acid cycle located in the cytoplasm, is composed of 405 amino acids and is a 2-oxoglutarate dehydrogenase involved in energy production and conversion.16,32
We produced SucB in recombinant form and evaluated the use of this antigen for the detection of C. burnetii antibodies in 3 ruminant species.
Materials and methods
Production of the recombinant protein in Escherichia coli
A C. burnetii Nine Mile RSA 493 phase I strain was produced in axenic medium in a BSL3 laboratory as described previously. 36 The bacteria were then centrifuged at 15,000 × g for 1 h at 4°C, and DNA was extracted (DNeasy blood and tissue kit; Qiagen) following the manufacturer’s instructions. According to NCBI (https://www.ncbi.nlm.nih.gov/) and to the prediction of SucB epitopes, 20 specific primers containing BamHI and EcoRI restriction enzyme sites were constructed and used to amplify the sucB gene (GenBank Q83BU7) in a 50-µL reaction mixture consisting of CoralLoad buffer 10× (Qiagen), 1.5 µL of 50 mM MgCl2, 1 µL of 10 mM dNTPs, 1 µL of each primer (10 μM; forward primer: 5′-TTGGATCCATGGCTATAGAAATTAAAGTACCTAC-3′, reverse primer: 5′-TTGAATTCACACTCCAAAATCATTCGAGCTG-3′), 100 ng of gDNA, and 0.25 μL of Taq DNA polymerase. PCR cycling conditions were PCR activation at 94°C for 3 min, denaturation at 94°C for 45 s, 35 cycles of annealing at 55°C for 30 s, and elongation at 72°C for 120 s, followed by a final elongation at 72°C for 10 min. The 1,215-bp product was examined in 1.5% agarose gel and digested for 3 h at 37°C with BamHI and EcoRI (Thermo Scientific).
After purification, the digestion product was ligated into a pSer expression vector previously digested with the same enzymes and used to transform competent E. coli BL21 C43 (DE3) cells plated in lysogeny broth (LB) agar medium with ampicillin.4,29 After colony confirmation by PCR assay, positive colonies were cultured in LB supplemented with ampicillin and induced with 1 mM isopropyl-D-1-thiogalactopyranoside (IPTG) during the mid-exponential phase. The insert presence as well as the in-frame orientation was further verified by Sanger sequencing of the DNA plasmid extracted from a positive clone. Bacteria were pelleted, lysed with lysozyme, sonicated, and then purified under denaturing conditions (1 M urea). The His-tagged fusion protein was purified in 3 affinity chromatography steps using an on-column nickel resin (HisPur Ni-NTA resin; Thermo Scientific). Purified eluates were pooled and dialyzed against 100 volumes of 0.1 M carbonate-bicarbonate buffer before being quantified (Bradford protein assay; Bio-Rad). SDS-PAGE, with Coomassie brilliant blue R250 staining, was used to separate and estimate the size of proteins within the eluate. Western blot was used to transfer the separated proteins to a nitrocellulose membrane, which was blocked with 5% casein in TBST (Tris-buffered saline 10× with 0.5% Tween 20) for 3 h at room temperature. A 6-His tag polyclonal antibody conjugated with horseradish peroxidase was used to react with recombinant SucB antigen in the western blot. A solution of 3,3′-diaminobenzidine tetrahydrochloride (MilliporeSigma) dissolved in 8 µM hydrogen peroxide in TBS (pH 7.6) was used for visualization.
Mass spectrometry analysis
Liquid chromatography–tandem mass spectrometry (LC-MS/MS) was performed as described previously.6,15 Briefly, an electrophoresis band was cut using a scalpel blade and destained in 50 mM ammonium bicarbonate buffer and 50% acetonitrile. Using 10 mM dithiothreitol and 50 mM iodoacetamide, the sample was reduced and alkylated. Protein digestion was performed using porcine trypsin (Merck) overnight at 37°C. Peptides were then separated using a BEH 130 C18 column (200 mm × 75 μm, particle size: 1. 7 μm; Waters) and analyzed by automated nanoflow reverse-phase liquid chromatography (nanoAcquity UPLC system, Q-TOF premier electrospray ionization tandem mass spectrometer; Waters). Data were obtained by spraying the sample at 3.4 kV capillary voltage in the Q-TOF detector, reading spectra from alternating scans at low and high collision energies (4–40 eV), and were queried in the reference C. burnetii RSA 493 strain database (UniProt Consortium, 9.10.2020 download, 1,824 entries, https://www.uniprot.org/). Identification was established if the sequence coverage achieved was ≥ 15% with an acceptable false-positive rate of 4%.
Development of the recombinant ELISA
Diluted in 0.1 M carbonate-bicarbonate buffer (pH 9.6), the recombinant protein (r-SucB) was used to coat (50 ng/well) the wells of 96-well plates (Nunc Maxisorp; MilliporeSigma) overnight at 4°C. After blocking with 2.5% bovine casein (200 µL/well), serum samples diluted 1:100 in PBS–1.25% casein (100 µL) were added to the wells and incubated at room temperature for 1 h. Following a wash step, the monoclonal antibodies anti-goat/sheep IgG peroxidase (Merck) or anti-bovine IgG peroxidase (Merck) were diluted (1:1,000 dilution in a volume of 100 µL/well) in the same buffer used for primary antibody incubation (45 min at room temperature). The reaction was developed with 100 µL of 3,3′,5,5′-tetramethylbenzidine (TMB) and stopped with 0.2 M H2SO4. Optical density was measured at 450 nm. To distinguish between positive and negative samples, a cutoff value of 0.45 was arbitrarily chosen (x¯ of negative sera + 4 SDs). These conditions were selected by testing different antigen amounts and different dilutions of positive and negative control sera (crisscross serial-dilution analysis). An initial panel of sera, consisting of 8 negative and 8 positive cattle, was used. A panel of sera belonging to clinically healthy ruminants (160 small ruminants, 120 cattle, 115 water buffalo) was thereafter chosen randomly for assessing the potential of r-SucB against a commercial ELISA (Q fever Ab test kit, catalog QFT1135T; Idexx). Serum samples were obtained from previous investigations performed in the Campania region, Italy.13,14 Before testing with our recombinant in-house ELISA, all sera were tested with the Idexx ELISA, and results compared.
Statistical analysis
Agreement was calculated for the results obtained with SucB and those obtained with the commercial ELISA. The Cohen κ coefficient was used to determine the level of agreement between the 2 tests (0.81–1.00 = almost perfect agreement; 0.61–0.80 = substantial agreement; 0.41–0.60 = moderate agreement; 0.21–0.40 = fair agreement; 0.01–0.20 = slight agreement; 0.00 = no agreement). We performed statistical analysis with MedCalc v.18.11.3 (MedCalc Software) and used GraphPad Prism 8 software (Dotmatics) to create Figures 3 and 4.
Results
The production of r-SucB in an E. coli heterologous system was successful as attested by the electrophoresis pattern of the gel stained with Coomassie blue (Fig. 1). Lysates from IPTG-induced E. coli were subjected to electrophoresis followed by western blot analysis to verify the presence of the 6-His tag in the protein of interest using a specific anti–6-His tag antibody. The 6-His tag was detected in the lysate, and the detected protein had the correct molecular weight (Fig. 2; Suppl. File 3). The protein identity was further verified using 2 different approaches. The correct in-frame insertion of the SucB product into the plasmid backbone was confirmed by Sanger sequencing. Moreover, the band observed in the SDS-PAGE gel was excised and verified by mass spectrometry, which detected a peptide consistent with the protein sequence available in the database (UniProt Consortium, 9.10.2020 download, 1,824 entries; Suppl. File 1). The peptide had identical physical and chemical properties and reached a coverage of 17.0% (higher than the limit of 0.15%; Suppl. File 1).
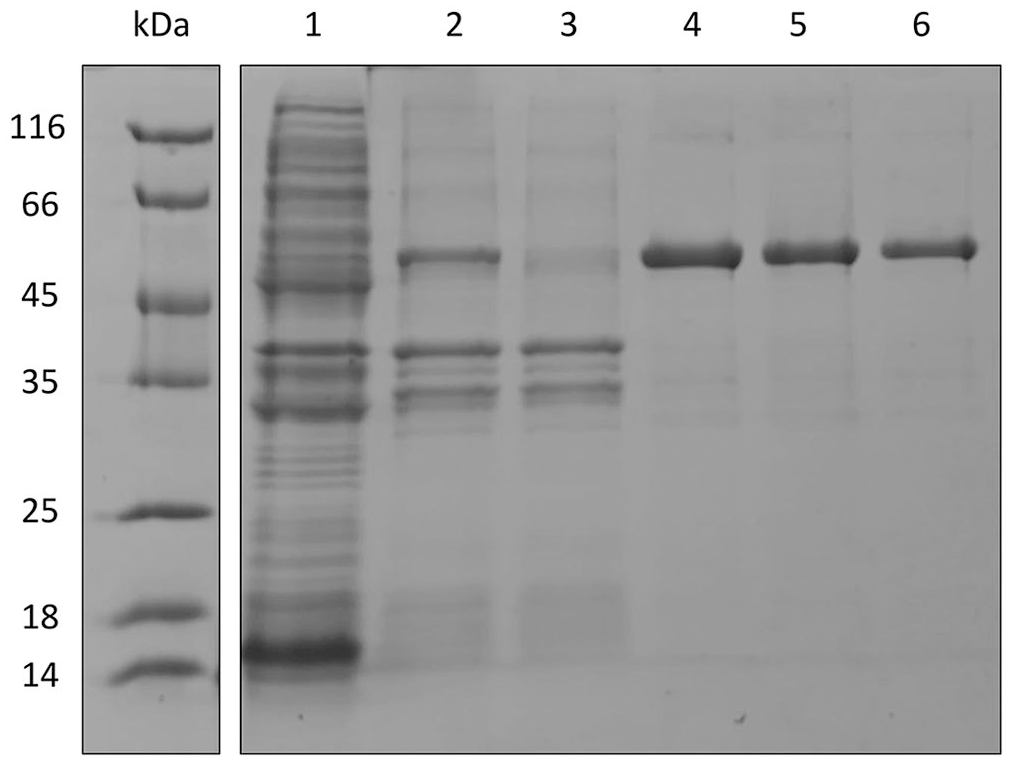
SDS-PAGE (Coomassie staining) analysis of recombinant SucB. Lanes: 1 = total lysate; 2 = total extract in 1 M urea; 3 = total extract in 1 M urea after incubation with nickel resin; 4–6 = SucB purified by affinity chromatography. All lanes have an overexpressed band of ~50 kDa, whose density decreased after resin incubation. Lanes 4–6 have a unique band of ~50 kDa, which corresponds to purified SucB. kDa = molecular weight ladder.
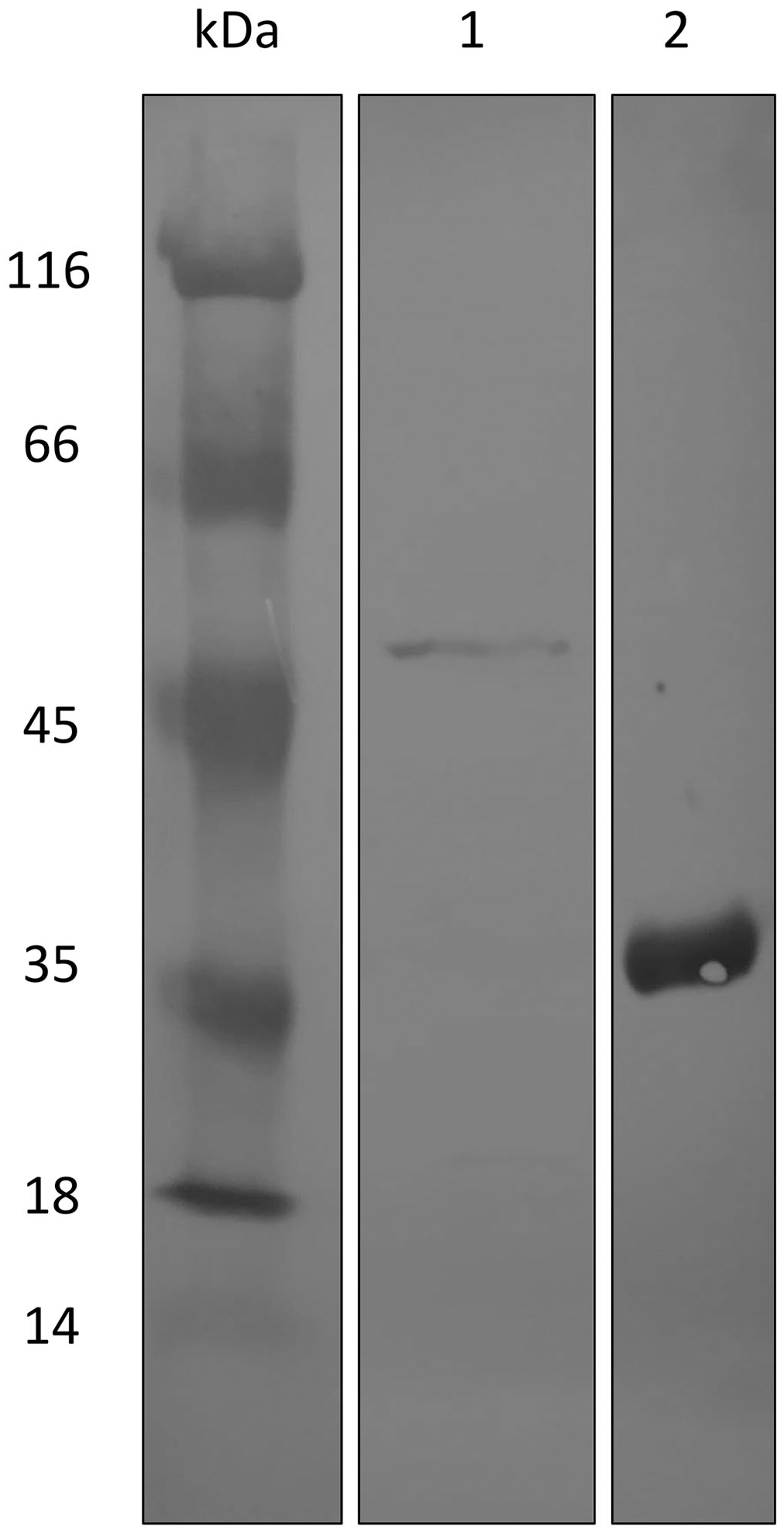
Western blot analysis of the 1 M urea extract produced by the transformed Escherichia coli. Lane 1 = a reactive band of ~50 kDa that corresponded to the protein of interest is highlighted with an anti–6-His tag antibody. Lane 2 = the actual reactivity of the antibody used was assessed using 6-His as a control. The full blot is available (Suppl. File 3). kDa = molecular weight ladder.
Given that the recombinant protein was poorly soluble, 1 M urea was used for extraction and purification. SDS-PAGE revealed a major band of the right size (molecular weight of ~50 kDa), as well as the absence of nonspecific bands corresponding to E. coli proteins (Fig. 1). The purity of the protein increased considerably in the purification eluates (E1–3). After pooling the 3 eluates, 0.62 mg/mL was obtained, according to the Bradford assay.
According to a serial dilution analysis (Fig. 3), the optimal conditions were to coat 50 ng and employ a 1:100 serum dilution. Using these criteria, an average of 0.23 (± 0.038) was obtained for the negative samples and 1.11 (± 0.33) for the positive samples in the first set of sera (8 positive and 8 negative cattle).
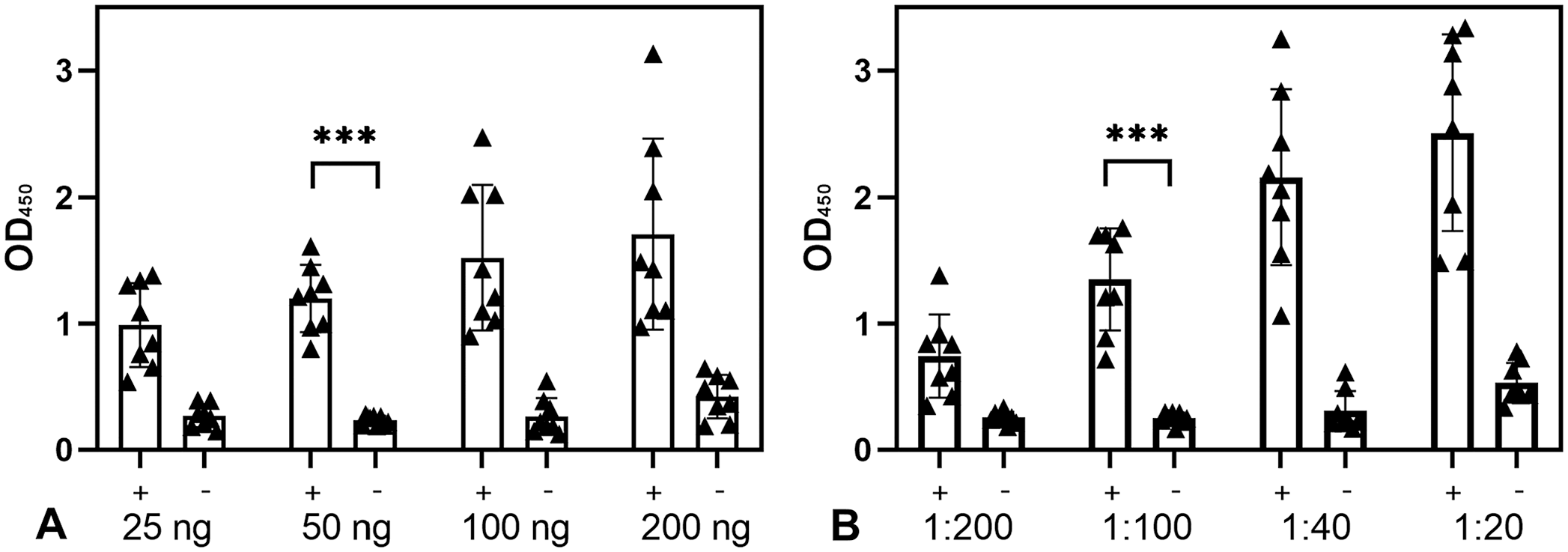
Optimal serum dilution and antigen concentration for the recombinant SucB ELISA.
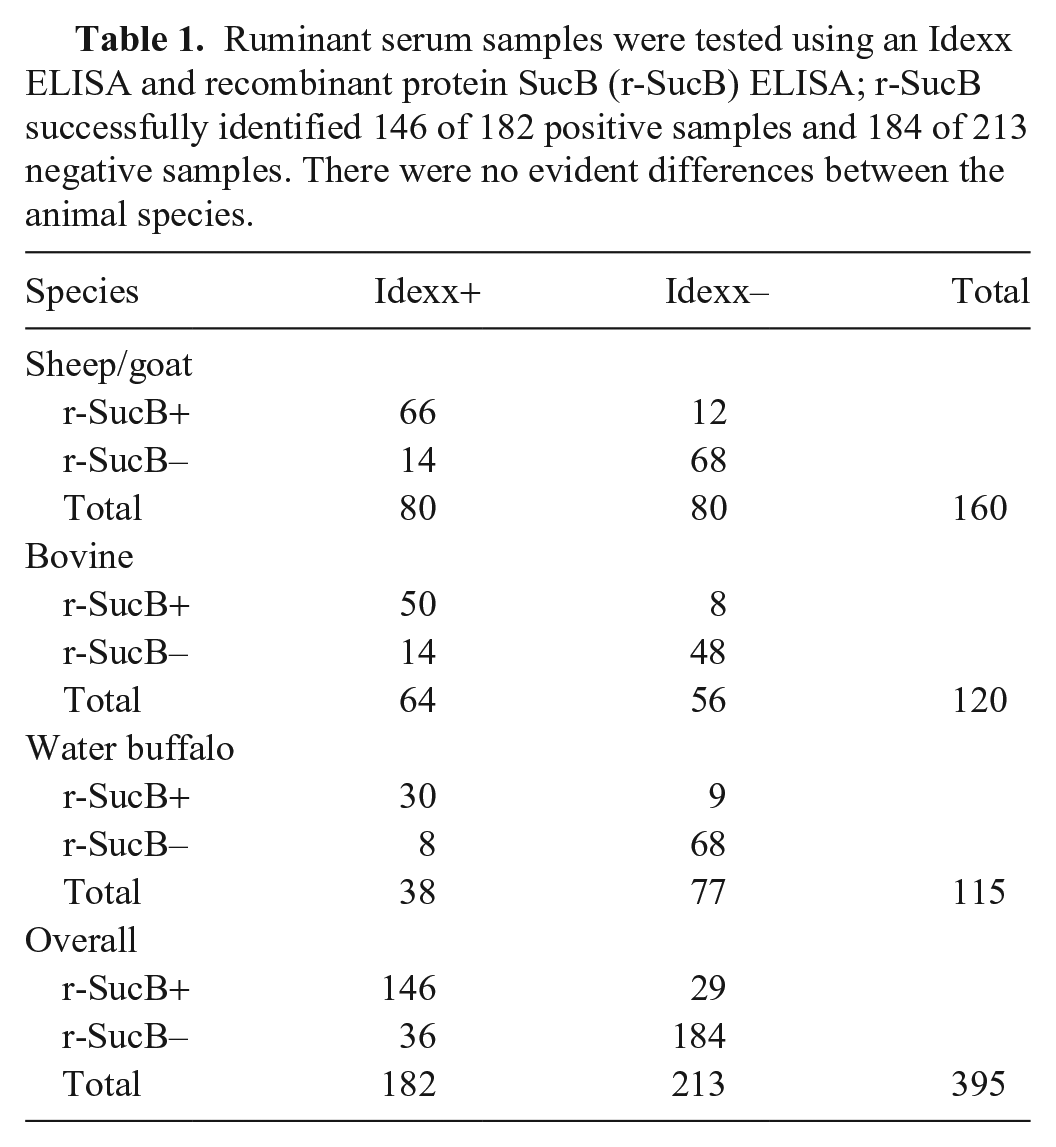
Our recombinant ELISA agreed with the Idexx ELISA results for 146 of 182 positive animals and 184 of 213 negative animals (Table 1). We detected 36 false-negatives and 29 false-positives, highlighting limitations of the performance of the recombinant test (83.5% agreement). The mean OD was 0.25 (range: 0.096–0.827) for negative samples and 0.99 (range: 0.12–3.24) for the negative and positive samples (Fig. 4; Suppl. File 4). The mode and median OD were 0.118 and 0.172 for the negative and 1.56 and 0.82 for the positive animals, respectively.
Ruminant serum samples were tested using an Idexx ELISA and recombinant protein SucB (r-SucB) ELISA; r-SucB successfully identified 146 of 182 positive samples and 184 of 213 negative samples. There were no evident differences between the animal species.
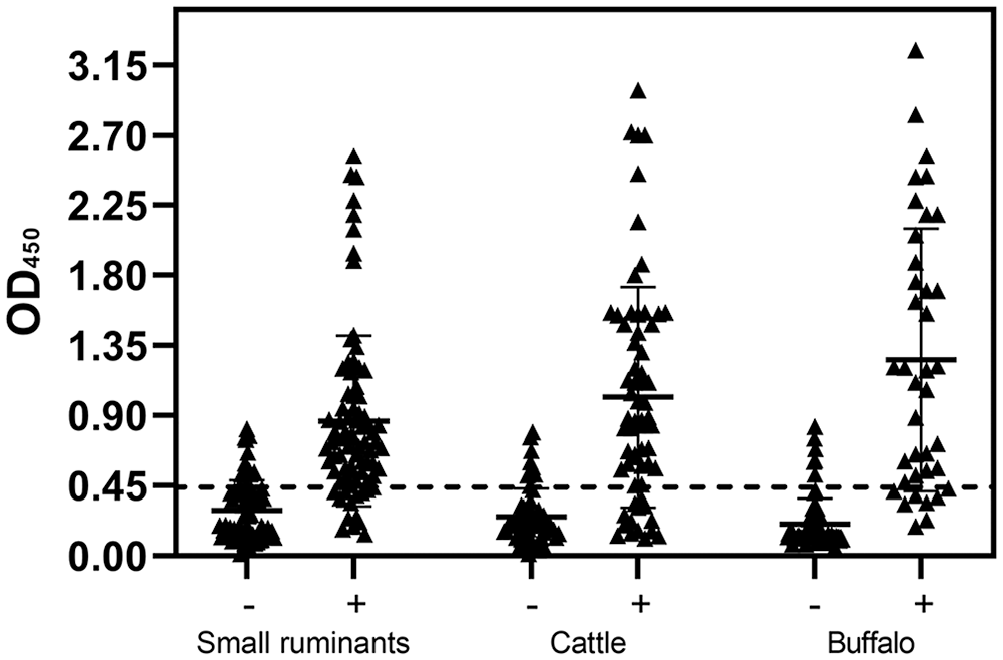
Recombinant SucB ELISA results from 160 small ruminants, 120 cattle, and 115 water buffalo, using optimal conditions (50 ng of antigen; serum dilution 1:100). The central bar and whiskers are the x¯ ± SD of the OD. The dashed line is the cutoff value.
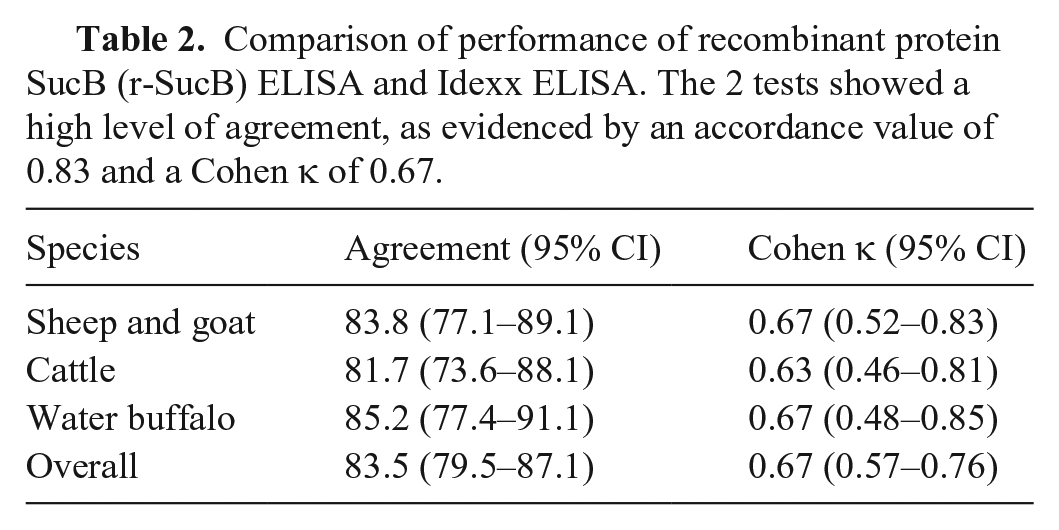
No major differences were found based on the species tested, although a lower number of false-negative results was found in small ruminants and a lower number of false-positive results was found in buffalo (Table 2). Substantial agreement was obtained in all species (Cohen κ was always >0.6, even if only slightly).
Comparison of performance of recombinant protein SucB (r-SucB) ELISA and Idexx ELISA. The 2 tests showed a high level of agreement, as evidenced by an accordance value of 0.83 and a Cohen κ of 0.67.
Discussion
We successfully produced the SucB antigen in a recombinant form, purified, and confirmed its identity by mass spectrometry. The 83.5% agreement obtained with this antigen for coating ELISA plates in comparison with a commercial ELISA is in line with those obtained in most similar studies.12,16,31 Several candidate antigens have been evaluated for the preparation of innovative ELISAs. The diagnostic performance of recombinant Com-1 was evaluated in bovine species and small ruminants, obtaining an accuracy of ~70%. 31 The performance of the recombinant Com-1 antigen was improved when it was used in synthetic form and for the preparation of a latex agglutination test (LAT).23,35 In the same species and with a similar approach, the performance of recombinant Ybgf was evaluated, obtaining sensitivity >80% and specificity >90%. 12 Other immunodominant antigens have been evaluated in ruminants obtaining good specificity but again low sensitivity. 26 To date, the recombinant antigen that has obtained the best performances is HspB (Se: 80–90%; Sp: 97%), which, however, has been evaluated exclusively in experimentally infected goats. 11
SucB has been identified as one of the main immunodominant antigens in humans and as one of the main candidates for future application in serodetection and vaccinology. SucB is among the 3 of 21 antigens identified to be useful for serodetection by protein microarray and immunostrip assay. 33 In a study including 33 Coxiella proteins, SucB also ranked in the top 3 for diagnostic performance, having reacted with ~70% of positive goat sera in a primary screen and with ~60% sensitivity and ~90% specificity using human sera. 26 The reason for the reduced diagnostic performance must first be sought in the complex immune response against C. burnetii, which is characterized in humans by an initial response against phase II antigens and a subsequent increase in antibodies against phase I.3,5
SucB is described in the literature as a phase II antigen, therefore, it should react mainly with antibodies produced in the acute form, not identifying, instead, antibodies produced in the chronic form. 16 Other causes must be sought in the performance of the reference ELISA used (described in the literature as suboptimal) and the failure to use reference sera for the evaluation of diagnostic performance. 32 The loss of specificity can be associated with cross-reactions with other pathogens as well as any post-translational modifications that E. coli, used for production, is unable to carry out and which could be fundamental for the definition of the epitopes. Even if our in-silico analysis (Suppl. File 2) ruled out any cross-reactions or high homologies with antigens from other microbes (including ruminant pathogens), it is necessary to consider the possibility of epitopes shared with microbes phylogenetically close to Coxiella and common in ruminants, such as Rickettsia spp., and Bartonella spp.17,24
Our assay is not ready for use in routine testing but requires additional testing by comparing the results with a larger number of sera, sera from experimentally infected animals, or reference sera. However, our work represents a further attempt to identify antibodies against Coxiella using a simple recombinant antigen as well as confirm the reactivity of SucB against most of the animals detected as seropositive with another test.
Supplemental Material
sj-pdf-1-vdi-10.1177_10406387231199964 – Supplemental material for Detection of Coxiella antibodies in ruminants using a SucB recombinant antigen
Supplemental material, sj-pdf-1-vdi-10.1177_10406387231199964 for Detection of Coxiella antibodies in ruminants using a SucB recombinant antigen by Gianmarco Ferrara, Barbara Colitti, Gabriela Flores-Ramirez, Ugo Pagnini, Giuseppe Iovane, Sergio Rosati and Serena Montagnaro in Journal of Veterinary Diagnostic Investigation
Footnotes
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
Our research was supported and funded by a PhD grant in the “innovative PhD with industrial characterization” included in the “2014-2020 national research and innovation operational program” (Italian Ministry of University and Research) and partially by the Scientific Grant Agency of the Ministry of Education of the Slovak Republic and the Slovak Academy of Sciences (VEGA- 2/0023/21), and the Slovak Research and Development Agency (APVV 19–0519).
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.