
Abstract
Based on publications reporting improvements in real-time PCR (rtPCR) performance, we compared protocols based on heat treatment or dilution followed by direct rtPCR to standard extraction and amplification methods for the detection of porcine reproductive and respiratory syndrome virus (PRRSV), influenza A virus (IAV), porcine epidemic diarrhea virus (PEDV), or Mycoplasma hyopneumoniae (MHP) in swine oral fluids (OFs). In part A, we subjected aliquots of positive OF samples to 1 of 4 protocols: protocol 1: heat (95°C × 30 min) followed by direct rtPCR; protocol 2: heat and cool (25°C × 20 min) followed by direct rtPCR; protocol 3: heat, cool, extraction, and rtPCR; protocol 4 (control): extraction and then rtPCR. In part B, positive OF samples were split into 3, diluted (D1 = 1:2 with Tris–borate–EDTA (TBE); D2 = 1:2 with negative OF; D3 = not diluted), and then tested by rtPCR using the best-performing protocol from part A (protocol 4). In part A, with occasional exceptions, heat treatment resulted in marked reduction in the detection of target and internal sample control (ISC) nucleic acids. In part B, sample dilution with TBE or OF produced no improvement in the detection of targets and ISCs. Thus, standard extraction and amplification methods provided superior detection of PRRSV, IAV, PEDV, and MHP nucleic acids in OFs.
Keywords
Conventional real-time PCR (rtPCR) requires nucleic acid extraction to recover nucleic acid templates for amplification from a sample. This process roughly consists of 4 phases: 1) cell lysis; 2) removal of membrane lipids, proteins, and other nucleic acids; 3) purification and/or binding; and 4) nucleic acid concentration 3 ; conventional extraction procedures based on commercial reagents and protocols involve up to 32 steps. 58 Conversely, direct rtPCR uses sample preparation methods that make nucleic acid rapidly available for amplification. The 3 general approaches to direct rtPCR are chemical (e.g., proteinase K, 23 alkaline solutions, 17 commercial lysis buffers 41 ); mechanical (e.g., sonication, 11 bead disruption 27 ); and physical (e.g., freeze–thaw, heat treatment30,40,53). Additionally, various treatments have been used in conjunction with heat treatment to improve the effect on direct rtPCR performance (e.g., sample dilution). Dilution of human oral fluid (OF) samples with PBS reportedly improved direct rtPCR detection rates 6 ; mucolytics were suggested to neutralize inhibitors 26 ; and dilution with Tris–borate–EDTA (TBE) was used to increase uniformity among samples, decrease viscosity, and facilitate direct rtPCR. 45 Similarly, in fecal and environmental samples, dilution of samples or nucleic acid extracts is a common pre-amplification step.1,4,34,36
Reportedly, direct reverse-transcription rtPCR (RT-rtPCR) of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) can be achieved by heat treatment of the sample, and/or the addition of a buffer, followed by amplification.5,24,46,50 Thus, “outstanding” RT-rtPCR performance for the detection of SARS-CoV-2 in human OF samples was reportedly achieved by diluting the sample with TBE and then heating at 95°C for 30 min. 45 In swine disease surveillance, testing of swine OFs is commonplace (e.g., ~460,000 tests were performed on OFs at 5 midwestern U.S. veterinary diagnostic laboratories during 2022; Trevisan G, Swine Disease Reporting System, pers. comm., 2023 Mar 10). For this reason, direct PCR for the detection of nucleic acid targets in swine OF specimens would be highly desirable both from the perspective of laboratory efficiency and lower cost.
Therefore, we compared various protocols for direct rtPCR versus standard extraction and amplification for rtPCR detection of porcine reproductive and respiratory syndrome virus 2 (PRRSV; Betaarterivirus suid 2), influenza A virus (IAV; Alphainfluenzavirus influenzae), porcine epidemic diarrhea virus (PEDV), and Mycoplasma hyopneumoniae (MHP) nucleic acids in swine OFs of known status.
Materials and methods
Experimental design
In part A, we evaluated the use of heat to achieve extraction-free rtPCR detection of PRRSV, IAV, PEDV, and MHP nucleic acids in OF samples. Samples known to contain PRRSV (n = 8), IAV (n = 8), PEDV (n = 8), or MHP (n = 8) were 2-fold serially diluted (neat, 1:2, 1:4, 1:8) with OF known to be free of PRRSV, IAV, PEDV, and MHP (n = 32 diluted samples per pathogen). Each diluted sample was then split into 4 aliquots, and each aliquot was randomized to 1 of 4 protocols: protocol 1: heat (95°C × 30 min) followed by direct rtPCR; protocol 2: heat, cool (25°C × 20 min), and then direct rtPCR; protocol 3: heat, cool, nucleic acid extraction, and rtPCR; protocol 4 (control): extraction and then rtPCR (Table 1).
Experimental design for part A (use of heat to achieve extraction-free real-time PCR detection).
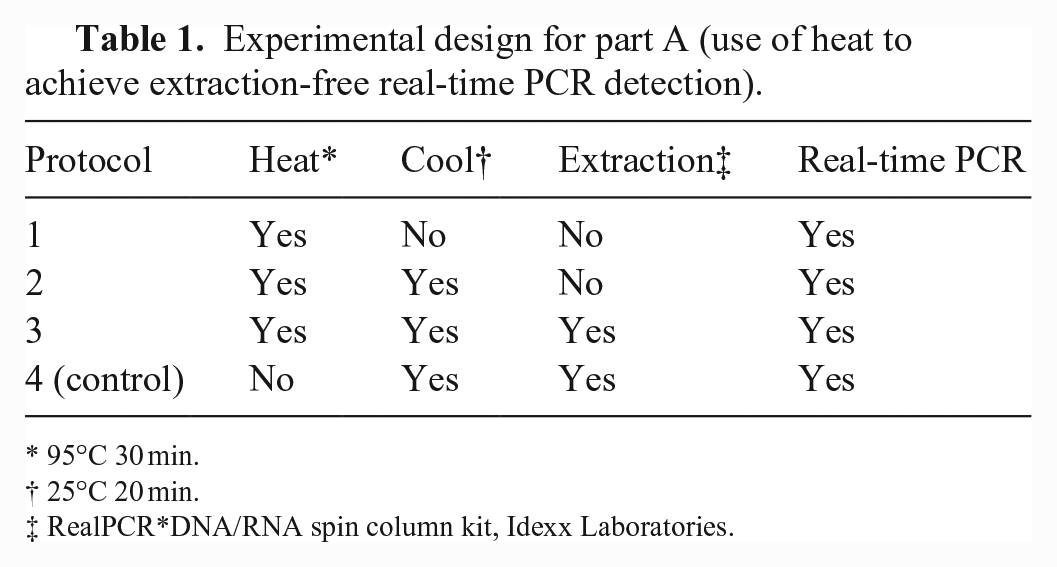
95°C 30 min.
25°C 20 min.
RealPCR*DNA/RNA spin column kit, Idexx Laboratories.
In part B, we assessed the effect of sample dilution with TBE on the detection of PRRSV, IAV, PEDV, and MHP using the best-performing rtPCR protocol from part A. OF samples known to contain PRRSV (n = 9), IAV (n = 10), PEDV (n = 10), or MHP (n = 10) were split into 3 aliquots and assigned to the following dilution protocols: D1 = diluted 1:2 with TBE; D2 = diluted 1:2 with OF free of PRRSV, IAV, PEDV, and MHP; D3 = undiluted. Aliquots (n = 30 aliquots per pathogen) were randomly ordered and then tested by rtPCR.
Oral fluid samples
OF samples used in parts A and B consisted of samples submitted to the Iowa State University Veterinary Diagnostic Laboratory (ISU-VDL; Ames, IA, USA) from commercial swine farms for routine testing or samples derived from pigs inoculated experimentally (Table 2).
Sample source and selection criteria for swine oral fluid samples used in part A (heat treatment) and part B (dilution with Tris–borate–EDTA).

dpi = days post-inoculation; IAV = influenza A virus; ISU-VDL = Iowa State University Veterinary Diagnostic Laboratory; MHP = Mycoplasma hyopneumoniae; PEDV = porcine epidemic diarrhea virus; PRRSV = porcine reproductive and respiratory syndrome virus.
Sample treatment
In part A, OF samples were thawed overnight at 4°C and 2-fold serially diluted (neat, 1:2, 1:4, 1:8) to a final volume of 800 µL using oral fluid known to be negative for PRRSV, IAV, PEDV, and MHP nucleic acids as the diluent. To compare treatment effects effectively, each dilution was split into 4 aliquots of 200 µL, and each aliquot was randomized (https://www.random.org) to 1 of 4 protocols involving heating and/or cooling and/or extraction followed by rtPCR (Table 2). For heating, samples were placed in a dry block heater (Standard dry block heater; VWR) and held at 95°C for 30 min. Cooling involved placing samples in an incubator at 25°C (12-140E; Quincy Lab) for 20 min. Temperature in the dry block was monitored with an alcohol thermometer (Fisherbrand; Thermo Fisher) inserted in a mock sample. Temperature in the incubator was monitored using an incubator bottle thermometer (Ertco; Thermo Fisher).
In part B, OF samples were thawed overnight at 4°C, split into 3 aliquots of 200 µL, and then completely randomized (https://www.random.org) to 1 of 3 dilution protocols: D1 = 1:2 dilution using TBE (89 mM Tris, 89 mM boric acid, 2 mM EDTA; Sigma-Aldrich); D2 = 1:2 dilution using OF known to be free of PRRSV, IAV, PEDV, and MHP; D3 = undiluted or “neat.” Aliquots were tested in consecutive random number order with the best-performing protocol from part A (protocol 4).
Nucleic acid extraction
Total nucleic acid extraction was performed (RealPCR DNA/RNA spin column kit; Idexx) following the instructions for the alternative protocol. In brief, 200 µL of OFs were added to 200 µL of a lysis working solution including 190 µL of lysis buffer, 5 µL of carrier RNA, and 5 µL of proteinase K, in addition to 2 µL of an internal positive control (RealPCR internal positive control; Idexx) in the case of IAV-positive OF samples. Binding conditions were adjusted with 200 µL of 96% ethanol (Thermo Fisher). Thereafter, 600 µL of lysate were transferred to a spin column and washed accordingly. Nucleic acids were eluted with 50 µL of elution water preheated to 70°C. Every nucleic extraction step included one positive and one negative extraction control. All extracts were directly subjected to rtPCR.
Real-time PCR assays
All rtPCR assays were performed using a commercial master mix and pathogen-specific target mixes (RealPCR PRRS types 1–2 RNA mix, RealPCR influenza A RNA mix, RealPCR PEDV RNA mix, RealPCR M. hyo DNA mix; Idexx). Target mixes for PRRSV and PEDV detect pathogen-specific RNA and an endogenous pig-specific RNA internal sample control (ISC). The IAV master mix targets IAV-specific RNA and an RNA external sample control spiked into the sample prior to heating (protocols 1–3) or extraction (protocol 4). The MHP master mix detects MHP-specific DNA and a pig endogenous DNA ISC. All rtPCR reactions were performed with 5 µL of extract or heat-treated OF sample using tubes compatible with the Mic qPCR system (Bio Molecular Systems) and a magnetic induction cycler (Mic qPCR cycler; Bio Molecular Systems) as directed by the reagent manufacturer: 50°C (15 min) for reverse transcription, denaturation at 95°C for 1 min, 45 cycles at 95°C for 15 s, followed by 60°C for 30 s for amplification. Every test included one positive and one negative extraction and amplification control. Results were analyzed using the Mic qPCR cycler software (v.2.10.4; Bio Molecular Systems) and reported as cycle threshold (Ct). According to the manufacturer’s recommendation, Cts < 40 were considered positive for all assays. Positive Cts were averaged as arithmetic means.
Data analysis
All analyses were performed using commercial statistical software (v.9.4; SAS Institute). A logistic regression model (Proc Genmod) was used to compare the number of positives among the 8 samples tested by each of the 4 PCR protocols (part A) with pathogen, protocol, dilution, and their interactions as input variables. A continuity correction was used to account for 8 negative results; 0.5 was added to the cumulative total for each protocol × dilution, and 1 was added to the total number of events (e.g., 0/8 became 0.5/9). Within pathogens, the slice statement was used to compare every protocol × dilution to protocol 4 (control) for both target and ISC. A linear mixed model (Proc Mixed) was used to analyze the effect of dilution on target and ISC Ct values in parts A and B. For part A, the analysis was performed using pathogen, protocol, dilution, and their interactions as fixed effects and aliquots within pathogen as a random effect. The slice statement was used to compare 2-fold dilutions to the “neat” aliquot for each pathogen and protocol. For part B, dilution protocol, target, and their interactions were used as fixed effects and aliquot nested within pathogen as a random effect. The slice statement was also used to compare dilution protocols to dilution protocol D3 (“neat” or undiluted). Analyses were considered significant at p ≤ 0.05 (parts A and B).
Results
In part A, using protocol 4 (control) as a comparison, heat treatment (protocols 1–3) negatively affected rtPCR detection of RNA viruses and their RNA ISCs both qualitatively (positive vs. negative) and quantitatively (Ct values; Table 3). With occasional exceptions, heat treatment (protocols 1–3) resulted in the complete loss of detectable target and ISC. A lesser effect was observed for MHP DNA and the ISC DNA, but target and ISC Cts were lower for protocol 4 in nearly all cases. With respect to serial dilution of samples, higher Cts were observed in target and ISC in samples diluted 1:2, 1:4, and 1:8 compared to neat samples. Although a pattern of increasing Cts was more evident in targets than ISCs, significant differences were infrequent.
Part A: effect of heat treatment on detection of target and internal sample control (ISC) detection by real-time PCR (rtPCR).
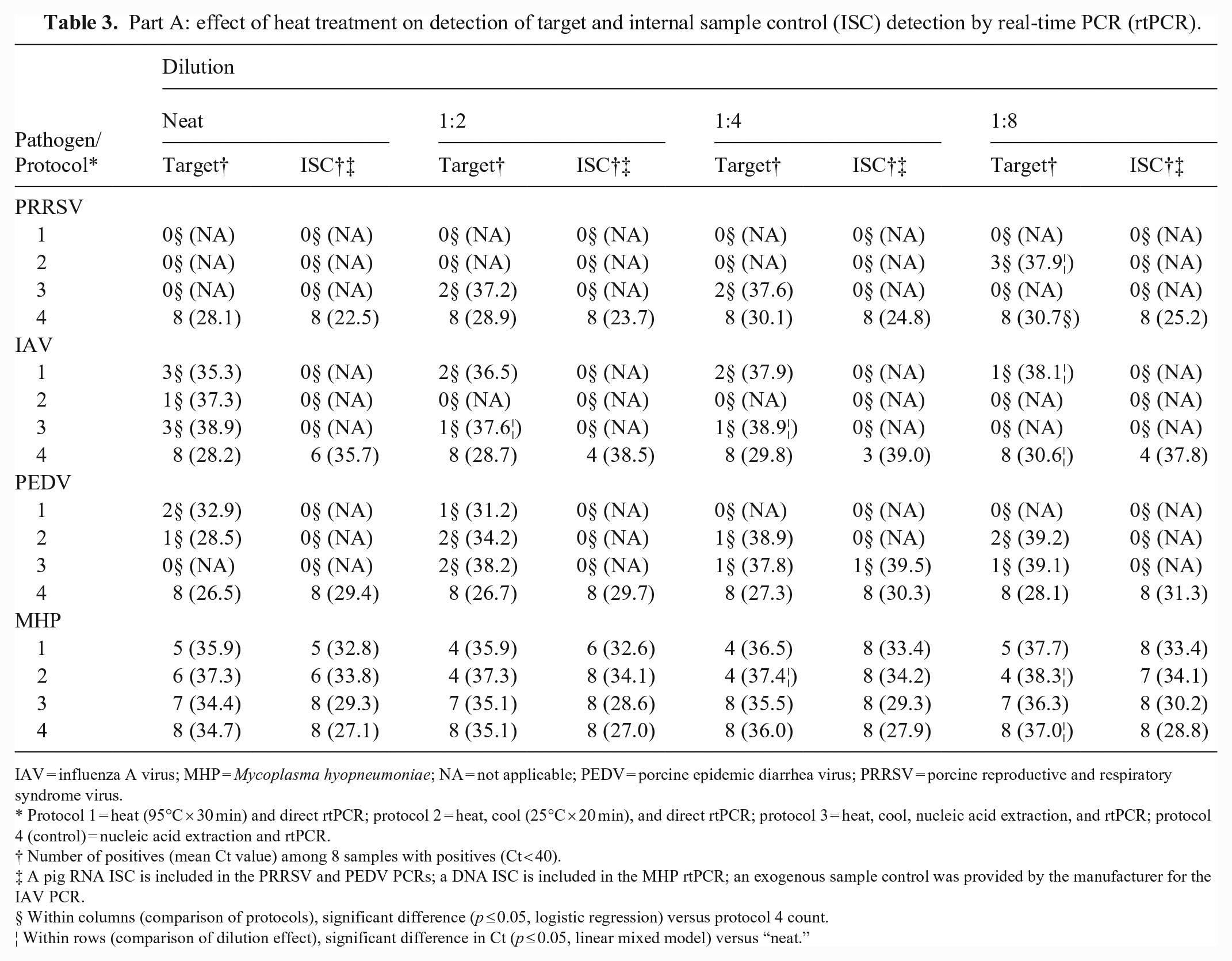
IAV = influenza A virus; MHP = Mycoplasma hyopneumoniae; NA = not applicable; PEDV = porcine epidemic diarrhea virus; PRRSV = porcine reproductive and respiratory syndrome virus.
Protocol 1 = heat (95°C × 30 min) and direct rtPCR; protocol 2 = heat, cool (25°C × 20 min), and direct rtPCR; protocol 3 = heat, cool, nucleic acid extraction, and rtPCR; protocol 4 (control) = nucleic acid extraction and rtPCR.
Number of positives (mean Ct value) among 8 samples with positives (Ct < 40).
A pig RNA ISC is included in the PRRSV and PEDV PCRs; a DNA ISC is included in the MHP rtPCR; an exogenous sample control was provided by the manufacturer for the IAV PCR.
Within columns (comparison of protocols), significant difference (p ≤ 0.05, logistic regression) versus protocol 4 count.
Within rows (comparison of dilution effect), significant difference in Ct (p ≤ 0.05, linear mixed model) versus “neat.”
In part B, the Ct results for targets and ISCs on undiluted (neat) samples were numerically equivalent (or better) than samples diluted with TBE or OF (Table 4). Cts for PRRSV and its ISC were significantly lower than samples diluted with OF or TBE.
Part B: effect of dilution on target and internal sample control (ISC) detection by real-time PCR.
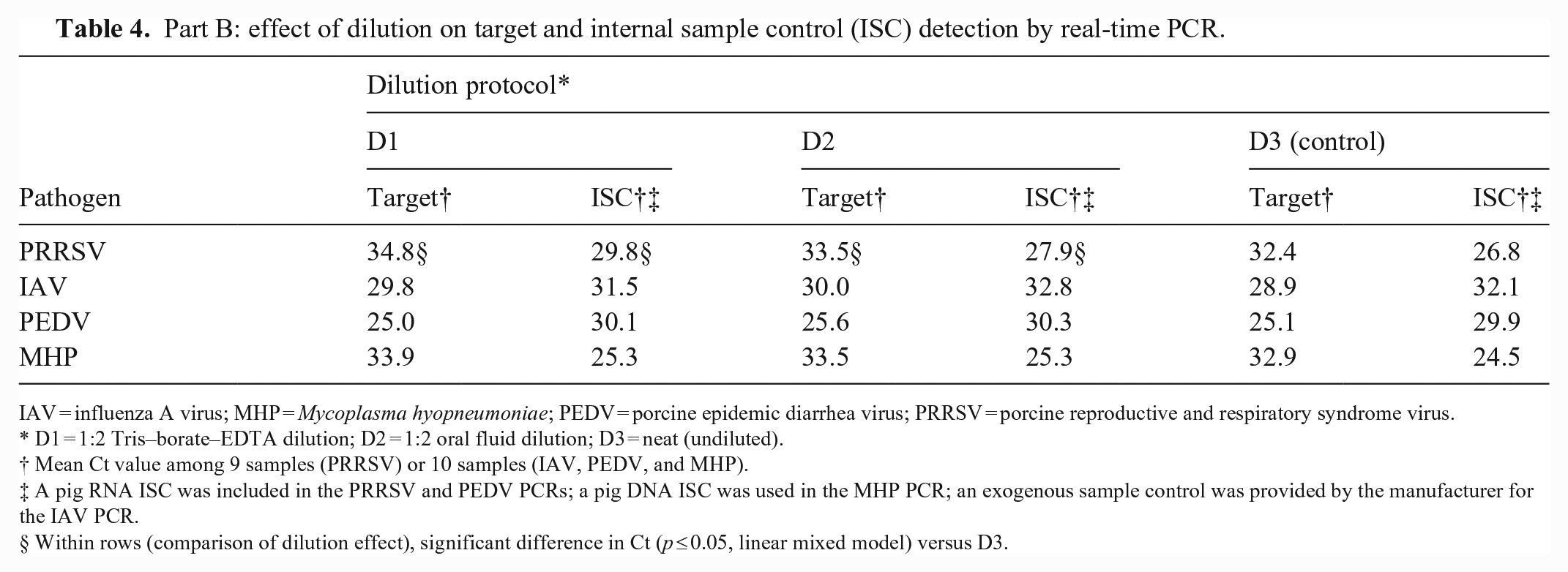
IAV = influenza A virus; MHP = Mycoplasma hyopneumoniae; PEDV = porcine epidemic diarrhea virus; PRRSV = porcine reproductive and respiratory syndrome virus.
D1 = 1:2 Tris–borate–EDTA dilution; D2 = 1:2 oral fluid dilution; D3 = neat (undiluted).
Mean Ct value among 9 samples (PRRSV) or 10 samples (IAV, PEDV, and MHP).
A pig RNA ISC was included in the PRRSV and PEDV PCRs; a pig DNA ISC was used in the MHP PCR; an exogenous sample control was provided by the manufacturer for the IAV PCR.
Within rows (comparison of dilution effect), significant difference in Ct (p ≤ 0.05, linear mixed model) versus D3.
Discussion
Direct PCR offers distinct advantages over conventional PCR (e.g., reduced processing time, lower cost, less sample handling, and less risk of contamination).35,57 Direct PCR was initially described for gel-based PCR testing for a variety of specimens (e.g., blood, 33 milk, 25 serum, 47 tissue 43 ), bacteria (e.g., Pseudomonas syringae, 51 Clostridium tyrobutyricum 25 ), fungi (e.g., Arthonia molendoi 62 ), and viruses (e.g., hepatitis C virus, 47 foot and mouth disease virus 48 ). After rtPCR became available, direct rtPCR was described for human OFs, 20 serum, 16 bacteria (e.g., Salmonella spp., 49 Campylobacter jejuni 9 ), and viruses (e.g., hepatitis B, 16 monkeypox 15 ).
In particular, the use of heat treatment is historically documented (e.g., for the detection of Mycobacterium tuberculosis DNA by PCR11,30,42,53). More recently, heat treatment has been used in the direct RT-rtPCR detection of SARS-CoV-2 RNA in human oral, oropharyngeal, and nasopharyngeal samples (Table 5), but our review of recent refereed publications (2020–2022) on SARS-CoV-2 showed extensive divergence in the temperatures (25–99°C) and times (3–60 min) reported. Further, some protocols combined heat treatment with pre- and post-treatments (e.g., addition of lysis solutions or a cool-down period). Thus, there is no consensus in the literature on the optimal conditions for direct PCR by heat treatment.
Procedures reported in 2020–2022 for SARS-CoV-2 direct reverse-transcription real-time PCR (RT-rtPCR) for human oral, oronasal, or oropharyngeal samples.
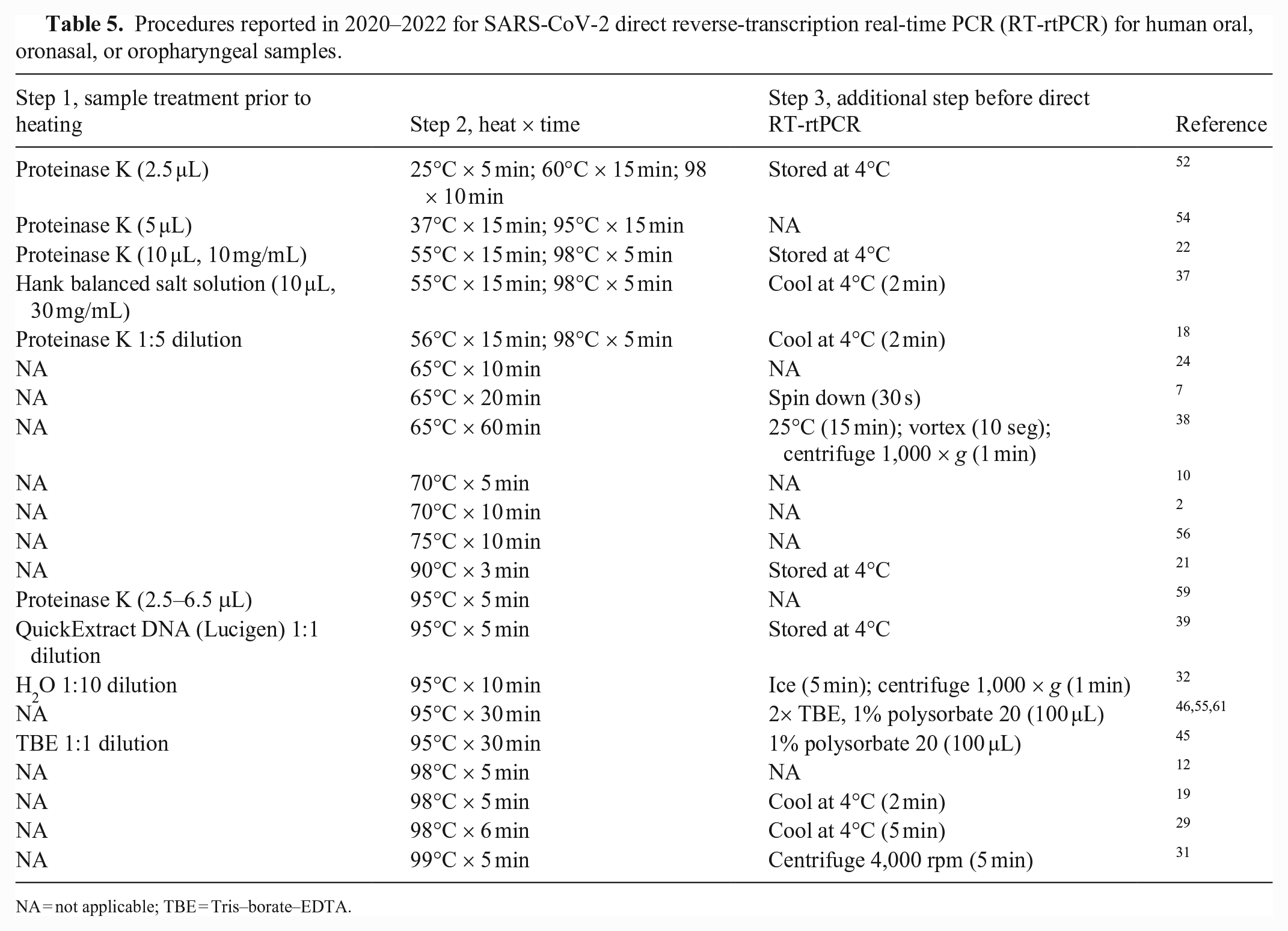
NA = not applicable; TBE = Tris–borate–EDTA.
Based on the reports of the successful detection of SARS-CoV-2 in oronasopharyngeal samples by heat treatment (95°C for 30 min),45,46,59 we compared heat treatment (95°C for 30 min) followed by direct RT-rtPCR to standard extraction and amplification procedures using swine OFs known to be positive for PRRSV, IAV, PEDV, and MHP. Heat treatment in part A (95°C × 30 min) was deleterious to the detection of PRRSV, IAV, PEDV, and MHP nucleic acids, even if the sample was cooled and/or nucleic acids were extracted using standard methods. Overall, heat treatment produced negative results or, in the case of a positive result, the Cts were higher than those produced by standard extraction and amplification methods (i.e., protocol 4, control). Further, RNA (i.e., PRRSV, IAV, PEDV, and their ISCs) was more affected by heat than DNA (i.e., MHP and its ISC). This difference can be explained by the presence of a highly reactive 2′-hydroxyl group in the RNA ribose unit, which makes RNA more susceptible to hydrolysis than DNA. 8 Studies based on agarose gel electrophoresis demonstrated that RNA is degraded at 95°C for 10 min 13 and DNA at 100–110°C for 5 min. 28 Thus, the nature of RNA and its thermolability are consistent with the low detection rates of viral RNA versus the higher resistance of DNA to heat and the higher detection rates of DNA under identical conditions.
In part B, a simple 2-fold dilution did not improve the detection of PRRSV, IAV, PEDV, MHP, or their ISCs. Diluting samples prior to amplification unavoidably reduces target concentration, with greater potential consequences (false-negatives) in the case of samples with low and medium target concentration. 14 Further, our findings coincide with other reports (e.g., dilution of swine OFs with nuclease-free water provided no improvement in PRRSV RT-rtPCR performance, 60 and dilution of human OFs with nuclease-free water did not correct for false-negatives and produced greater variability in Cts 61 ).
We found that standard extraction and amplification procedures provided far better rtPCR performance than heat treatment or sample dilution using well-characterized OF samples collected from infected individual animals (i.e., not spiked samples). When juxtaposed with the direct PCR protocols reported in the refereed literature, our results suggest that greater attention should be given to experimental design; full factorial designs are rarely possible because of cost and logistical complexity, but reports of experimental PCR protocols should necessarily include comparisons to standard extraction and amplification protocols.
Footnotes
Declaration of conflicting interests
The authors declared no conflicts of interest with respect to their authorship and/or the publication of this manuscript, with the exception that Jeffrey Zimmerman serves as a consultant to Idexx Laboratories. The terms of the consulting arrangement have been reviewed and approved by Iowa State University in accordance with its conflict of interest policies.
Funding
Our project was partially funded by Pork Checkoff Funds distributed by the National Pork Board (NBP 20-171, SHIC 20-157; Des Moines, IA, USA). PCR reagents were kindly provided by Idexx Laboratories. In addition, Betsy Armenta-Leyva received support from Consejo Nacional de Ciencia y Tecnología (México), Becas CONACYT-Regional Noroeste 2020.