
Abstract
Neorickettsia risticii, an obligate intracellular bacterium, is the causative agent of Potomac horse fever (PHF). Diagnosis of PHF is based on demonstration of serum antibodies, isolation of N. risticii, and/or detection of nucleic acid by a PCR assay. An existing real-time PCR assay targeting the N. risticii 16S rRNA has been validated using blood samples from horses with colitis, and snails; to our knowledge, the performance of the assay for other sample types has not been reported. We describe here a modification of the 16S rRNA gene assay by the addition of a set of primers and probe targeting the N. risticii p51 gene to form a duplex assay. We validated the new assay using diagnostic specimens from 56 horses with suspected PHF. The assay consistently detected down to 5 copies of synthetic targets, and did not show any cross-reaction with common equine enteric pathogens. Although we did not establish the diagnostic sensitivity and specificity of the duplex assay, results for both gene targets were in complete agreement, with the exception of 4 fecal samples that tested positive for the 16S rRNA gene only. Further analysis indicated that testing of fecal samples using our 16S rRNA gene assay alone can produce a false-positive result.
Neorickettsia risticii (formerly Ehrlichia risticii), an obligate intracellular gram-negative bacterium, is the causative agent of Potomac horse fever (PHF).6,10 PHF, which is a common form of equine colitis in endemic areas, is seasonal and occurs commonly during late spring to early fall with higher incidence observed in animals residing near bodies of water. 11 The disease has been reported from North and South America and Europe.1,14,15,18 Within the United States, the disease occurs most frequently in northeastern, mid-Atlantic, and western regions. 17
A 2020 study reported a new species of Neorickettsia, named N. findlayensis, causing an illness similar to PHF in horses in eastern Ontario, Canada. 19 N. findlayensis is phylogenetically more divergent from N. risticii than N. sennetsu, the causative agent of human Sennetsu neorickettsiosis.5,19 To date, horses infected with N. findlayensis have been reported from Ohio in the United States, and in Alberta, Quebec, and Ontario in Canada.1,3,19
Neorickettsiae are intracellular endosymbionts of digenean trematodes that have a complex life cycle involving sexual reproduction in a vertebrate definitive host and asexual reproduction in an intermediate host, typically an aquatic snail. 9 Neorickettsiae are maintained through the life cycle of digenean trematodes by vertical transmission. 9 N. risticii is maintained in several trematode species that parasitize insectivorous birds and/or bats and use river snails as the first intermediate host and aquatic insects as the second intermediate host. 8 Horses, considered accidental hosts, acquire infection horizontally when they ingest N. risticii–infected trematode metacercariae within aquatic insects or free-living trematode cercariae in bodies of water. 20 After ingestion, N. risticii replicates in colonic epithelial cells, tissue macrophages, mast cells, and blood monocytes. 4 Clinical manifestations of PHF in naturally infected horses include diarrhea, fever, anorexia, depression, colic, laminitis, and abortion in pregnant mares. 2
Diagnosis of PHF is based on demonstration of serum antibodies, isolation of N. risticii from whole blood, and detection of nucleic acid by a PCR assay.13,16 Serologic assays are associated with a high rate of false-positive results given the poor correlation of serum antibody titers with infection. 12 Neorickettsiae require cell culture for isolation. Therefore, identification of N. risticii antigen or nucleic acid is required to support a definitive and timely disease diagnosis. A real-time PCR (rtPCR) assay targeting the N. risticii 16S rRNA gene and validated using blood samples from horses with infectious colitis and snails has been described. 16 To our knowledge, the performance of the assay for other sample types, particularly feces, which is a common specimen type collected for investigation of diarrhea in horses, has not been reported.
We describe here a modification of the 16S rRNA gene assay by addition of a set of primers and probe targeting the N. risticii p51 gene for improved detection of N. risticii from diagnostic specimens. The p51 gene in neorickettsiae encodes for P51, a surface-exposed 51-kDa antigenic protein. 7 Although the sequence of p51 is known to be strain variable in external loops 2 and 4, the conserved regions within the gene serve as useful targets for development of PHF tests. 7 Primers and probe sequences used for amplification and detection of the N. risticii 16S rRNA gene were described previously, except that the probe was labeled with reporter dye, hexachlorofluorescein (HEX) at the 5′-end and black hole quencher-1 (BHQ1) at the 3′-end (Biosearch Technologies). 16 Using an online primers-and-probe design tool (https://www.genscript.com/tools/real-time-pcr-taqman-primer-design-tool), we designed primers (forward PHF-F2: 5′-GTGCAGTTGTAATGGCTCGT-3′; reverse PHF-R2: 5′-ACACGAAATTCACAGCATCG-3′) and a probe (PHF-Probe2: 5′-FAM-TGACCCAAACGGGATCGATAACTG-BHQ1-3′) to amplify a 65-bp fragment of the p51 gene. Primer specificity was verified using NCBI Primer-BLAST (https://www.ncbi.nlm.nih.gov/tools/primer-blast/). Furthermore, we aligned the selected 65-bp target region of the p51 gene to sequences in GenBank using NCBI BLAST (https://blast.ncbi.nlm.nih.gov/Blast.cgi) and visualized using the Multiple Sequence Alignment Viewer application (Suppl. Fig. 1). The selected region is identical in 10 strains of N. risticii for which sequences were available in GenBank, indicating that the region is highly conserved in N. risticii. The primer-binding regions had ≥2 nucleotide mismatches with other Neorickettsia spp. The probe-binding region had 5 nucleotide mismatches with N. sennetsu strains and 7 nucleotide mismatches with N. findlayensis and Neorickettsia sp. SF agent (Neorickettsia species denoted as the fluke Stellantchasmus falcatus agent), indicating a high in silico sequence specificity of the selected primers and probe for N. risticii.
PCR reactions were performed as singleplex assays targeting either the p51 gene or 16S rRNA gene, or a duplex assay targeting both genes. The PCR reactions consisted of a master mix (AccuStart II PCR ToughMix; Quantbio), forward primers, reverse primers, and probe in a 20-µL mix, and 5 µL of template. All primers and the PHF-probe2 were used in a final concentration of 400 nM; the final concentration of the 16S rRNA gene probe was 80 nM. Thermocycling conditions consisted of initial denaturation at 94°C for 3 min, followed by 45 cycles of 94°C for 15 s, 57°C for 25 s, and 72°C for 30 s (CFX96 touch real-time PCR detection system; Bio-Rad).
DNA extraction from whole blood samples was performed (QIAamp DNA mini kit; Qiagen) following the manufacturer’s recommendations. DNA was extracted from feces and intestinal mucosal scrapings (QIAamp DNA stool kit; Qiagen) and from spleen and intestinal tissues (Wizard genomic DNA purification kit; Promega) following the manufacturer-recommended protocol for animal tissues. DNA was extracted from formalin-fixed, paraffin-embedded (FFPE) tissues (DNA extract all reagents kit; Thermo Fisher) after deparaffinizing the tissue in xylene.
We first assessed the analytical performances of the 16S rRNA gene and the p51 gene PCR assays independently as singleplex assays (Fig. 1A, 1B). DNA extracted from the spleen of a horse diagnosed with PHF was used as the positive control. The histopathologic changes in the intestine of the horse were consistent with PHF, and the spleen had tested positive by the N. risticii 16S rRNA gene assay performed at the University of Cornell Animal Health Diagnostic Center (Ithaca, NY, USA). 16 Serial 10-fold dilutions of the DNA were tested in triplicate wells, and amplification efficiencies were calculated from the slope of the curve. Amplification efficiencies of 90.8% and 103% were observed for the 16S rRNA gene and p51 gene assays, respectively. The linearity (R) measures whether the amplification efficacy is the same for different starting template concentrations. The R2 value of 0.995 for the 16S rRNA assay and 0.999 for the p51 gene assay indicated a high correlation between the Ct value and the starting DNA concentration. Next, we assessed the analytical performance of the 16S rRNA gene and p51 gene duplex assay (Fig. 1C). Analysis of a serial 10-fold dilution series of the positive control in triplicate wells resulted in amplification efficiency of 98.0% with linearity R2 = 0.999 for the 16S rRNA gene target, and efficiency of 100% with linearity R2 = 0.999 for the p51 gene target. The data indicated little interference in amplification of both targets when the 16S rRNA gene and p51 gene assays were performed as a duplex assay.
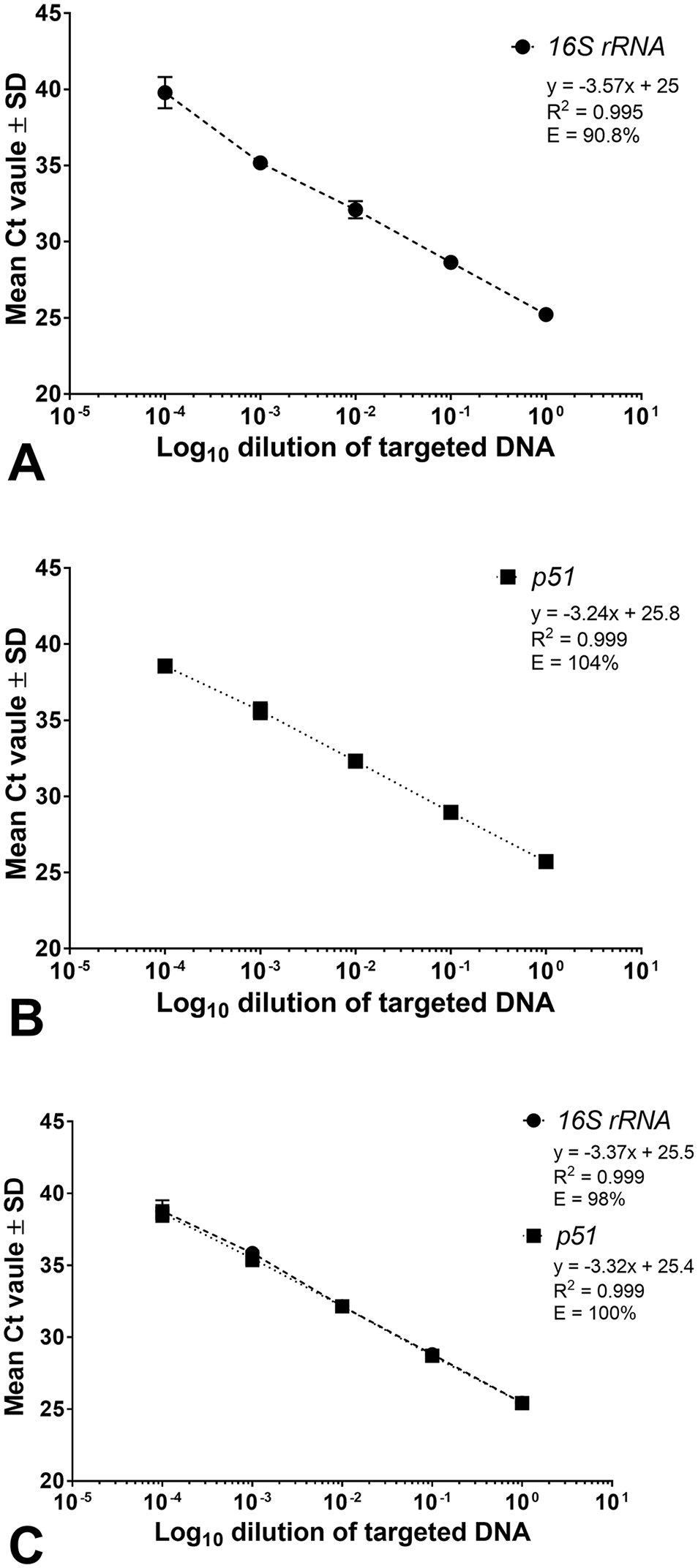
Standard curves of the
To determine repeatability of the duplex assay, 3 dilutions (10−1, 10−3, 10−4) of DNA extracted from the positive spleen were tested in 5 replicates over 6 d. Intra-assay and inter-assay variations for each target were determined by calculating the CV from the replicates. For both intra-assay and inter-assay, SDs and CVs for both targets were <0.1% and <2.5%, respectively.
To determine limit of detection for the duplex assay, we tested 125-bp long synthetic double-stranded DNA fragments (gBlocks gene fragments; Integrated DNA Technologies) consisting of the 16S rRNA gene or p51 gene PCR targets. The reactions included 10-fold serial dilutions containing 500,000 to 5 copies of the synthetic targets per reaction in addition to lower theoretical copy numbers of 2.5, 1.25, 0.5, per reaction in triplicate. The assay consistently detected down to 5 copies of both targets in 3 of 3 replicates (Suppl. Fig. 2). The dynamic range, which is the range of input template over which a reaction is linear, was at least 5-log dilutions for both of the targets, although we did not determine the upper limit of the dynamic range. In addition, amplification efficiency of 94.9% with linearity R2 = 0.999, and efficiency of 102% with linearity R2 = 0.998, were achieved for the 16S rRNA gene and p51 gene gBlock targets, respectively. The efficiency of amplification and linearity for both synthetic targets were similar to those determined using the positive diagnostic specimen (Fig. 1C; Suppl. Fig. 2).
Next, we assessed the performance of the duplex assay by testing 81 diagnostic specimens from 56 horses with suspected PHF (Table 1; Suppl. Table 1). The tested samples included paired blood and fecal samples from 24 horses. Of the 56 horses, 23 (41%) animals tested positive for both the 16S rRNA gene and the p51 gene. Of the 24 paired samples, both blood and fecal samples from 11 (46%) animals tested positive for both targets. Only blood, but not the fecal sample, from one animal was positive for both targets. Most PCR-positive horses had typical clinical manifestations of PHF, including fever, diarrhea, and laminitis (Suppl. Table 1). Notably, fecal samples from 4 animals tested positive for the 16S rRNA gene with Ct values of 36, 35, 37, and 37, but were negative for the p51 gene (Suppl. Table 1). Blood samples from these 4 animals were negative for both targets. Of the 4 horses, 2 had fever and diarrhea, 1 had fever, and no clinical history was available for another. To exclude the possibility of nonspecific amplification as a result of primer combinations in the duplex assay, we tested all 4 fecal samples with discrepant PCR results using singleplex assays. All 4 samples tested positive by the 16S rRNA gene assay with similar Ct values and were negative by the p51 gene assay. In addition, attempts to amplify the N-terminal region of the Neorickettsia strain–specific antigen 3 gene (Ssa3) from the 4 fecal samples using the primer sets (840–1 and 840–2) described previously 19 failed to yield expected PCR amplicons. Collectively, these findings indicated that testing of fecal samples with the 16S rRNA gene assay alone can produce a false-positive result, and testing of paired blood and fecal samples using the duplex assay improves assay specificity and sensitivity.
Diagnostic performance of the Neorickettsia risticii 16S rRNA gene and p51 gene duplex real-time PCR assay on samples from 56 horses with suspected Potomac horse fever.

FFPE = formalin-fixed, paraffin-embedded; Paired samples = both blood and fecal samples from 24 animals.
We assessed the specificity of the N. risticii duplex assay by testing for bacterial enteric pathogens encountered commonly in horses. Isolates of Rhodococcus equi ATCC 6939, Salmonella enterica Typhimurium ATCC 14028, Clostridium perfringens ATCC 3626, C. perfringens ATCC 27324, and Cryptosporidium parvum (waterborne) tested negative for both the N. risticii 16S rRNA and p51 gene targets by the duplex assay. In addition, DNA from N. findlayensis strain Fin17 tested positive for 16S rRNA gene and negative for the p51 gene. This was not unexpected given that the 16S rRNA PCR target region was identical between N. risticii and N. findlayensis except for 2 nucleotide mismatches in the forward primer–binding region of the N. findlayensis 16S rRNA gene.
A limitation of our study is that we did not attempt to isolate N. risticii from diagnostic specimens and therefore, we could not determine the diagnostic sensitivity and specificity of the duplex PCR assay. Most diagnostic laboratories use PCR for the detection of neorickettsiae because isolation of neorickettsiae in cell culture may take several days to weeks, making isolation less practical. The sensitivity of a nested PCR assay for the detection of N. risticii in blood and fecal samples has been found comparable to culture. 13
A 2022 study reported development and validation of SYBR green–based rtPCR assays targeting the Neorickettsia Ssa2 gene for differential detection of N. risticii and N. findlayensis using clinical isolates and blood samples from confirmed and suspected PHF cases. 3 However, the utility of these assays for testing of other specimen types, including fecal samples, needs to be validated. Additional studies are needed to understand geographic distribution and prevalence of Neorickettsia spp. causing Potomac horse fever.
Supplemental Material
sj-pdf-1-vdi-10.1177_10406387221135184 – Supplemental material for Improved molecular detection of Neorickettsia risticii with a duplex real-time PCR assay in the diagnosis of Potomac horse fever
Supplemental material, sj-pdf-1-vdi-10.1177_10406387221135184 for Improved molecular detection of Neorickettsia risticii with a duplex real-time PCR assay in the diagnosis of Potomac horse fever by Nagaraja R. Thirumalapura, Julia Livengood, John Beeby, Weihua Wang, Erin L. Goodrich, Laura B. Goodman, Erdal Erol and Deepanker Tewari in Journal of Veterinary Diagnostic Investigation
Footnotes
Acknowledgements
We thank Dr. Susan Bender from the University of Pennsylvania, School of Veterinary Medicine for providing diagnostic specimens from PHF cases. We also thank Joy Hecht from the CDC Rickettsial Isolate Reference Collection for providing the Neorickettsia findlayensis strain Fin17 DNA (CRIRC NFI001).
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.