
Abstract
Vector-borne pathogens, such as Bourbon virus (BRBV), Heartland virus (HRTV), West Nile virus (WNV), and Trypanosoma cruzi (TCZ) are a great threat to public health and animal health. We developed a panel of TaqMan real-time PCR assays for pathogen surveillance. PCR targets were selected based on nucleic acid sequences deposited in GenBank. Primers and probes were either designed de novo or selected from publications. The coverages and specificities of the primers and probes were extensively evaluated by performing BLAST searches. Synthetic DNA or RNA fragments (gBlocks) were used as PCR templates in initial assay development and PCR positive controls in subsequent assay validation. For operational efficiency, the same thermocycling profile was used in BRBV, HRTV, and WNV reverse-transcription quantitative PCR (RT-qPCR) assays, and a similar thermocycling profile without the initial reverse-transcription step was used in TCZ qPCR. The assays were optimized by titrating primer and probe concentrations. The analytical sensitivities were 100, 100, 10, and 10 copies of gBlock per reaction for BRBV (Cq = 36.0 ± 0.7), HRTV (Cq = 36.6 ± 0.9), WNV (Cq = 35.5 ± 0.4), and TCZ (Cq = 38.8 ± 0.3), respectively. PCR sensitivities for vector genomic DNA or RNA spiked with gBlock reached 100, 100, 10, and 10 copies per reaction for BRBV, HRTV, WNV, and TCZ, respectively. PCR specificity evaluated against a panel of non-target pathogens showed no significant cross-reactivity. Our BRBV, HRTV, WNV, and TCZ PCR panel could support epidemiologic studies and pathogen surveillance.
Vector-borne diseases caused by bacteria, parasites, and viruses account for >17% of all infectious diseases worldwide (https://www.who.int/news-room/fact-sheets/detail/vector-borne-diseases). Disease transmission involves the ingestion of a pathogen by the vector during a blood meal from an infected host, replication of the pathogen in the vector, and infection of new hosts during subsequent bites. 31 The prevalence of vector-borne diseases is heavily influenced by the geographic and seasonal distribution of vectors, as well as demographic, environmental, and social factors.8,20 The common symptoms among people with vector-borne diseases are fever, chills, headache, myalgias, arthralgias, and anorexia; severe cases can be fatal.
Of the known vector-borne diseases, anaplasmosis, ehrlichiosis, Lyme disease, tularemia, and West Nile fever have been prevalent for decades, whereas Bourbon virus (BRBV; Orthomyxoviridae, Thogotovirus), Heartland virus (HRTV; Phenuiviridae, Bandavirus, Heartland bandavirus), and Trypanosome cruzi (TCZ) are newly emerging health threats in the midwestern United States. BRBV was first detected in 2014 in Bourbon County, Kansas. The patient, who had presented with tick bites, developed symptoms and eventually died of cardiac arrest. 10 In 2017, another fatal case of BRBV infection occurred in Missouri. A retrospective analysis of 39,096 ticks collected during the spring and summer of 2013 from northwestern Missouri detected the virus in 3 pools of Amblyomma americanum ticks. 24
HRTV is also transmitted by A. americanum. In 2009, 2 individuals from northwestern Missouri were diagnosed with HRTV infection. The patients were hospitalized with fever, fatigue, anorexia, diarrhea, leukopenia, and thrombocytopenia. 15 A subsequent epidemiologic study detected neutralizing antibodies in northern raccoons, horses, white-tailed deer, dogs, and Virginia opossums. 4
West Nile virus (WNV; Flaviviridae, Flavivirus, West Nile virus) emerged in New York City in 1999. WNV infection has been the leading cause of mosquito-borne disease in the continental United States. About 20% of infected individuals have mild symptoms, such as fever, and <1% of patients develop neurologic disease. The virus is maintained in nature between birds and mosquitoes (mostly, the Culex species: C. pipiens, C. tarsalis, C. quinquefasciatus, which serve as the main vector for the virus). The virus can also infect cats, dogs, horses, rabbits, skunks, squirrels, and alligators. 17
TCZ is a flagellated protozoon responsible for Chagas disease (American trypanosomiasis), the top parasitic disease in the Americas. The parasite is carried by hematophagous triatomine bugs, primarily Rhodnius prolixus, in their guts and transmitted via contamination of the bite site or intact mucous membranes by bug feces. Woodrats, raccoons, opossums, armadillos, and skunks serve as carriers of TCZ. Acute Chagas disease causes flu-like symptoms, but chronic infection may result in heart failure and severe gastrointestinal illness. 3 In 2017, the first autochthonous TCZ infection case in Missouri was diagnosed in a blood donor through screening. 32
Active surveillance of vector-borne pathogens is a key step in the successful prevention and control of vector-borne diseases. The surveillance program usually includes monitoring the introduction of vectors, changes in the density of vectors, and the presence of pathogens. PCR has been used widely to detect pathogens in vectors and hosts. Although various PCR protocols have been published, it is laborious to use different PCR methods in a laboratory to test a panel of target pathogens. In addition, some published primers are not able to cover all prevalent strains of the pathogens. To streamline laboratory operations and improve sample throughput, we developed and validated a uniform reverse-transcription quantitative PCR (RT-qPCR) protocol for the detection of BRBV, HRTV, and WNV, and a qPCR for TCZ, in their primary vectors because these pathogens are associated with emerging and re-emerging infections in the midwestern United States.
Materials and methods
Primers, probes, and gBlocks
Our BRBV and HRTV RT-qPCRs amplify a 100-bp fragment of BRBV segment 1 (GenBank KU708253.1, nucleotides 520–619) and the HRTV S fragment (GenBank KJ740146.1, nt 309–408), respectively (Table 1). Our WNV RT-qPCR amplifies a 72-bp fragment of the envelope gene of WNV (GenBank KM212944.1, nt 194–265). Our TCZ qPCR amplifies a 166-bp region of the satellite DNA (GenBank AY520036.1, nt 26–191). The specificity and coverage of the primers and probes were determined using BLAST (https://blast.ncbi.nlm.nih.gov/Blast.cgi) to search the NCBI nucleotide database. Synthetic DNA or RNA fragments (gBlocks) were used as PCR templates and positive controls in initial assay development and subsequent assay validation, respectively. All primers, probes, and gBlocks were synthesized by Integrated DNA Technologies.
Primers, probes, and gBlocks used in our study.
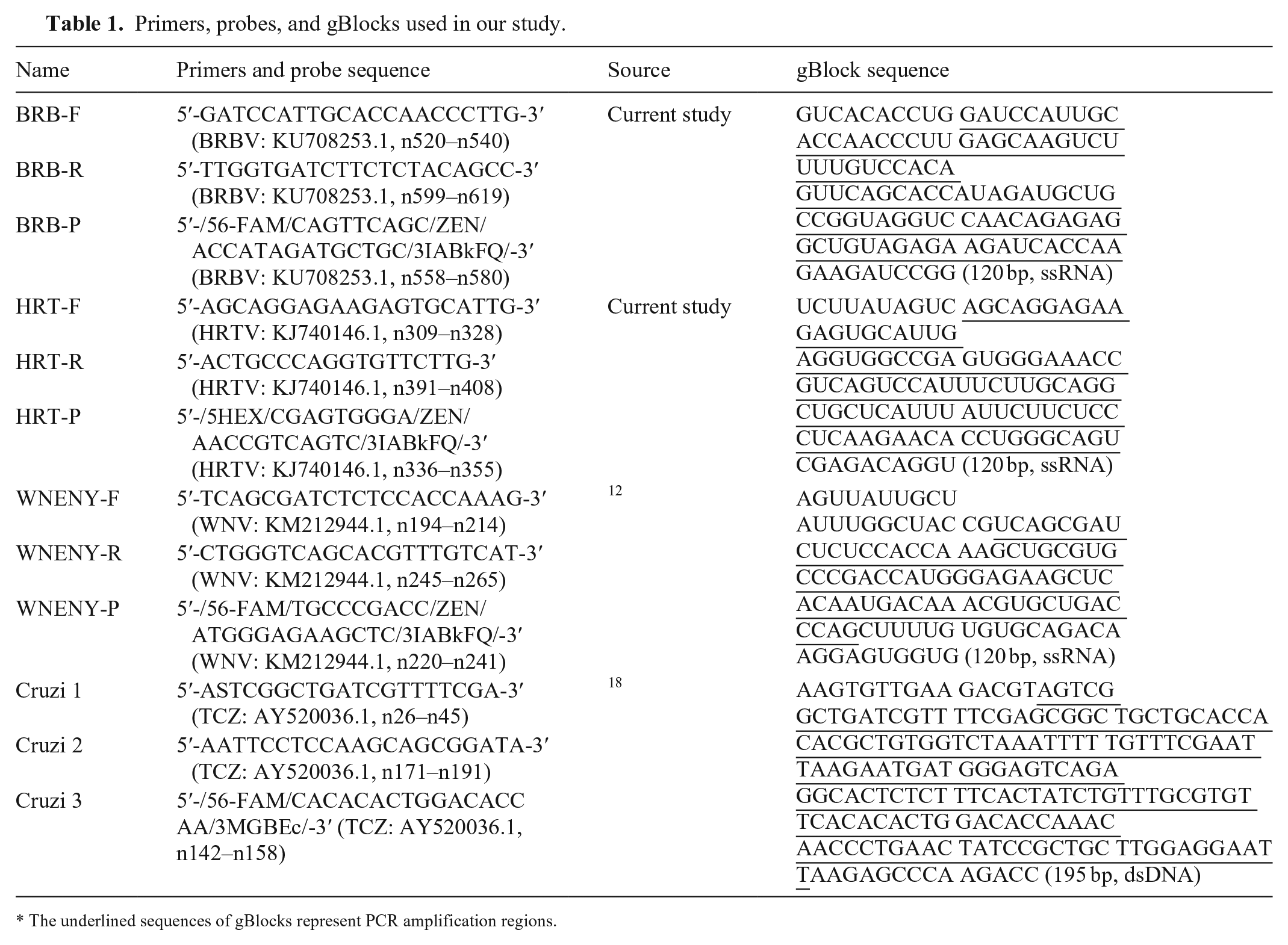
The underlined sequences of gBlocks represent PCR amplification regions.
Pathogen genomic DNA or RNA
Genomic RNA of BRBV Original strain, HRTV MO-4 strain, and WNV Bird 114, CO 1862, and BA 80900-4 strains, as well as gDNA of TCZ G, Dm28c, and CL strains, were obtained from the BEI Resources Repository, National Institute of Allergy and Infectious Diseases (Rockville, MD). The abovementioned gDNA and gRNA, in addition to gBlocks, were used to validate the RT-qPCR or qPCR assays. The RNA extracted from infected Vero cells contained cellular RNA, as notified by the BEI Resources Repository.
Vector DNA and RNA
A. americanum ticks, C. tarsalis mosquitoes, and R. prolixus bugs were also procured from the BEI Resources Repository. Pooled (4 or 5) ticks or mosquitoes were placed into a 1.5-mL sterile microcentrifuge tube containing Lysing Matrix H beads (MP Biomedicals) in 500 µL of Dulbecco PBS (DPBS; Gibco) and homogenized (Bead Mill 24 homogenizer; Thermo Fisher) for 90 s. RNA was extracted from 100 µL of the homogenate (NucleoSpin RNA mini kit; Macherey-Nagel) according to the manufacturer’s instructions. For DNA extraction from R. prolixus, the protocol is similar except only 1 adult bug was homogenized in lysis buffer T1 containing Lysing Matrix H beads, and 180 µL of homogenate was used for DNA extraction with the NucleoSpin tissue mini kit. The concentration and purity (A260/280 and A260/230) of extracted DNA and RNA were measured (NanoPhotometer; Implen).
PCR optimization
PCR assays were optimized by adjusting primer and probe concentrations under the same or similar thermocycling conditions. PCR reactions were carried out on an ABI 7500 fast real-time PCR system (Applied Biosystems). BRBV, HRTV, and WNV RT-qPCR assays were conducted (AgPath-ID one-step RT-PCR reagents; Life Technologies) according to the manufacturer’s instructions. A 25-µL reaction consisted of 12.5 µL of 2× RT-PCR master mix, 5 µL of template RNA, 2 µL of each primer, 2 µL of probe, and 1.5 µL of double-distilled water (ddH2O). The thermocycling profile was as follows: 45°C for 10 min, 95°C for 10 min, and 40 cycles of 95°C for 15 s and 60°C for 45 s. TCZ qPCR was performed (QuantiTect probe PCR master mix; Qiagen) according to the manufacturer’s instructions. The reaction volume was also 25 µL, consisting of 12.5 µL of 2× PCR master mix, 5 µL of template DNA, 2 µL of each primer, 2 µL of probe, and 1.5 µL of ddH2O. The thermocycling profile was as follows: 95°C for 10 min, and 45 cycles of 95°C for 15 s and 60°C for 45 s. The gBlock DNA or RNA were included as positive amplification controls, and nuclease-free water served as negative amplification controls.
The following primer and probe concentrations: 5 µM, 10 µM, 15 µM, and 20 µM were evaluated in initial optimization. To avoid nonspecific reaction and reduce the probability of primer–dimer formation, the lowest primer and probe concentrations giving strong fluorescent signals were chosen for each assay.
Analytical sensitivity and specificity
Ten-fold serial dilutions of the gBlocks at concentrations ranging from 2 × 10−1 to 2 × 106 copies/µL were subjected to PCR analysis, equivalent to 100–107 copies per reaction. All PCR assays were repeated 3 times. A standard curve for each assay was constructed by plotting the mean cycle quantification (Cq) values against the template concentrations. Serial dilutions of gRNA (Original strain of BRBV, MO-4 strain of HRTV, BA 80900-4 strain of WNV) or gDNA (Dm28c strain of TCZ) were also subjected to PCR for sensitivity analysis.
Specificity of the assays was determined by testing against the following vector-borne pathogens: 4 bacterial species transmitted by ticks (Ehrlichia sp., Anaplasma sp., Rickettsia rickettsii, Borrelia burgdorferi), 2 parasites transmitted by tsetse flies and sand flies (Leishmania major, Trypanosoma brucei), and 3 RNA viruses transmitted by mosquitoes (yellow fever virus, dengue virus, and Zika virus). DNA of the 4 tick-borne bacterial species was extracted from positive clinical samples confirmed in the University of Missouri, Veterinary Medical Diagnostic Laboratory (Columbia, MO, USA). Other gDNA and gRNA for specificity testing were obtained from the BEI Resources Repository.
PCR performance on vector DNA or RNA spiked with gBlock and gRNA or gDNA
A. americanum RNA was spiked with serial dilutions of BRBV gBlock, HRTV gBlock, BRBV gRNA, or HRTV gRNA. Similarly, C. tarsalis RNA was spiked with WNV gBlock or WNV gRNA; R. prolixus DNA was spiked with TCZ gBlock or TCZ gDNA. The spiked vector RNA or DNA was subjected to RT-qPCR or qPCR analysis, respectively.
Statistical analysis
A nonparametric test, the Wilcoxon–Mann–Whitney test, was used to analyze differences in Cq values obtained from PCR analysis between pure gBlock, gRNA or gDNA, and vector RNA or DNA spiked with gBlock or gRNA or gDNA (Cq = mean ± SD, n = 3, p ≤ 0.05).
Results
Coverage and specificity of primers and probes
There were 20 BRBV sequences (5 segment 1, and 3 segments 2–6) and 24 HRTV sequences (5 segment Land M, 13 segment S) in the NCBI nt database. We designed primers and probes to achieve maximum (100%) coverage of the sequences of each pathogen. BLAST indicated that BRBV primers/probe set matched all 5 segment 1 sequences (KU708253.1, MH880287.1, MK453529.1, KY825740.1, KY825741.1) and the HRTV primers/probe set matched all 13 S segment sequences (JX005842.1, JX005843.1, NC_024496.1, KJ740146.1, KC466555.1–KC466563.1). Although a Batken virus (Orthomyxoviridae, Thogotovirus) strain (GenBank MN987250.1) and a Dhori virus (Orthomyxoviridae, Thogotovirus, Dhori thogotovirus) strain (GenBank MT628422.1) showed some similarities to the BRBV amplification region, there were sufficient mismatches in the forward primers, reverse primers, and the probe (5-3-1 nt and 3-3-1 nt, respectively) to prevent nonspecific amplification. With regard to HRTV primers, the most closely related sequences are those of the severe fever and thrombocytopenia syndrome virus (Phenuiviridae, Bandavirus, Dabie bandavirus). However, there were still 8 nt mismatches in the forward primer. WNV primers and probe were adopted from a 2000 study. 12 The primers and probe covered >500 WNV sequences in the NCBI nt database. The most closely related viral sequence was from a Japanese encephalitis virus (Flaviviridae, Flavivirus) strain, but there were 9 nt mismatches in the WNV forward primer. The TCZ primers and probe, 18 also demonstrated broad coverage for >100 TCZ sequences deposited in the NCBI nt database. However, there was only 1 nt mismatch between the TCZ forward primer and 2 Trichomonas vaginalis sequences (RefSeq XM_001294727.1, XM_001291938.1).
Optimal primer and probe concentrations
To improve laboratory efficiency, an identical thermocycling profile for all 3 RT-qPCR assays, and a similar profile for the qPCR assay without the initial reverse-transcription step, were used to optimize primer and probe concentrations. For each assay, a concentration of gBlocks giving a moderate Cq value of ~25 was used as PCR template and mixed with primers and probe at different concentrations, including 5 µM, 10 µM, 15 µM, and 20 µM. The lowest primer and probe concentrations resulting in a strong fluorescent signal for a given assay were chosen as the optimal concentrations, which were as follows: 10 µM primer and probe for BRBV, HRTV, and WNV assays; and 15 µM primer and probe for the TCZ assay.
Assay specificity
The BRBV, HRTV, and WNV RT-qPCRs were analyzed for cross-reactions with both RNA and DNA derived from non-target pathogens; the TCZ qPCR was only evaluated against DNA extracted from non-target pathogens. BRBV RT-qPCR and HRTV RT-qPCR did not show cross-reaction with each other. Neither of these assays reacted with the RNA of 3 mosquito-borne pathogens (yellow fever virus, dengue virus, and Zika virus), DNA of 4 tick-borne pathogens (Ehrlichia sp., Anaplasma sp., R. rickettsii, B. burgdorferi), or DNA of 3 parasite species (TCZ, T. brucei, L. major). WNV RT-qPCR did not react with any of the non-target pathogens described above. TCZ qPCR did not react with the above non-target DNA, except that a weak signal was detected when high concentrations (>0.6 ng/µL) of L. major DNA were subjected to analysis.
Assay sensitivity
When using gBlocks as PCR templates, the detection limits of the assays were 102 copies/reaction for BRBV and HRTV, and 101 for WNV and TCZ. One order below these concentrations resulted in non-detection or inconsistent detection. Standard curves were constructed using Cq values at 6 concentrations (102–107 copies/reaction for BRBV and HRTV and 101–106 copies/reaction for WNV and TCZ). The RT-qPCR and qPCR amplifications were highly efficient, as indicated by the efficiency (slope) and correlation coefficient (R2) values (Fig. 1).
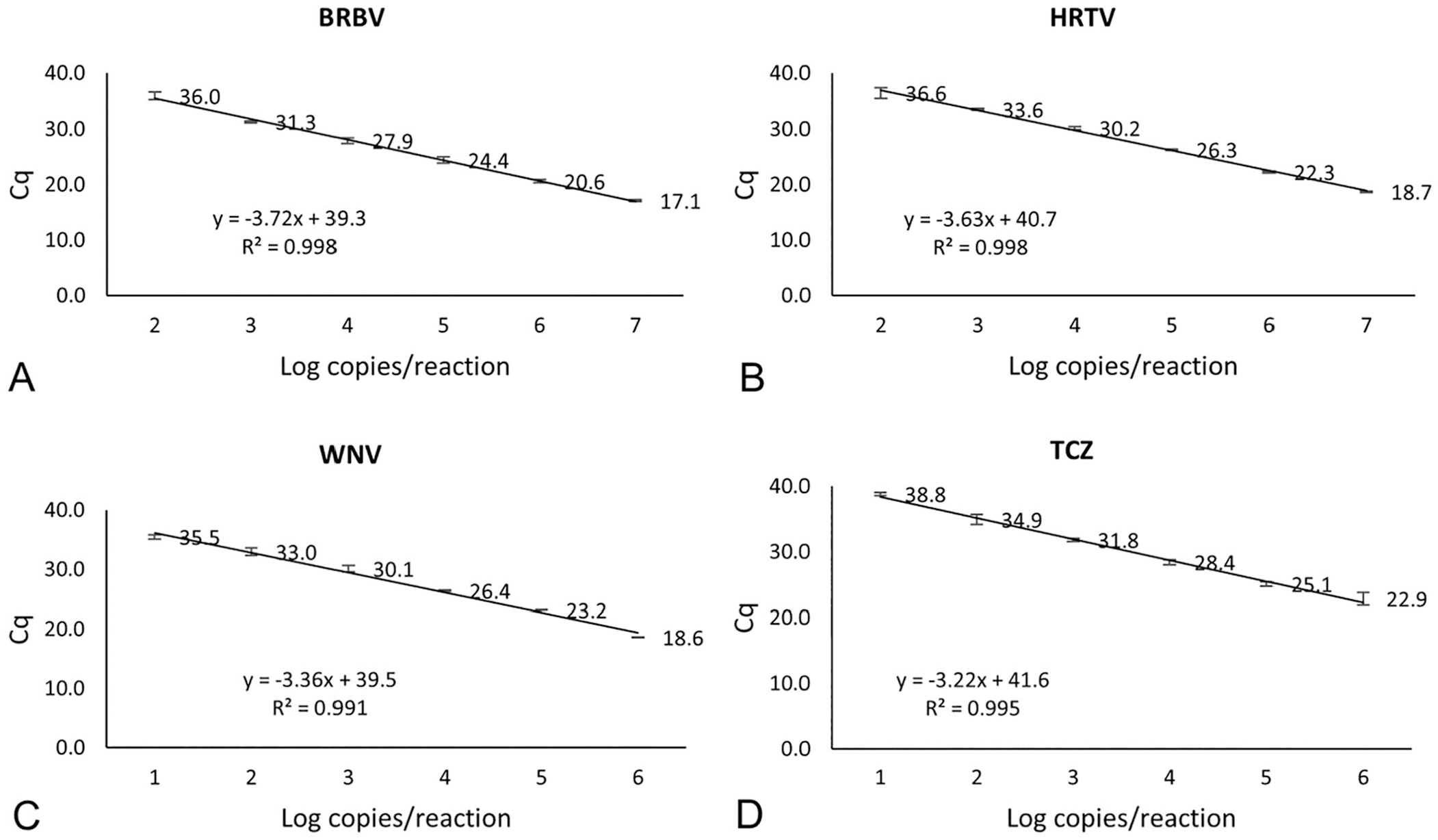
Standard curves of
When gRNA and gDNA were tested, the detection limits of BRBV, HRTV, WNV, and TCZ assays were at 104, 105, 105, and 109 dilutions, respectively (Fig. 2). The undiluted TCZ gDNA concentration was 35.6 ng/μL. At the 109 dilution, 5 μL of TCZ gDNA per reaction corresponded to 0.178 fg of genome of TCZ Dm28c strain or 0.003 parasites per reaction based on the genome size of 53.3 mbp of the strain. 2 Because BRBV, HRTV, and WNV gRNA preparations contained unknown amounts of host cellular RNA and carrier RNA, the detection limits could not be converted to gene copies per reaction. However, the PCR assays showed comparable amplification efficiency (slope) and correlation coefficient (R2) values to that of the assays using gBlocks as PCR templates.
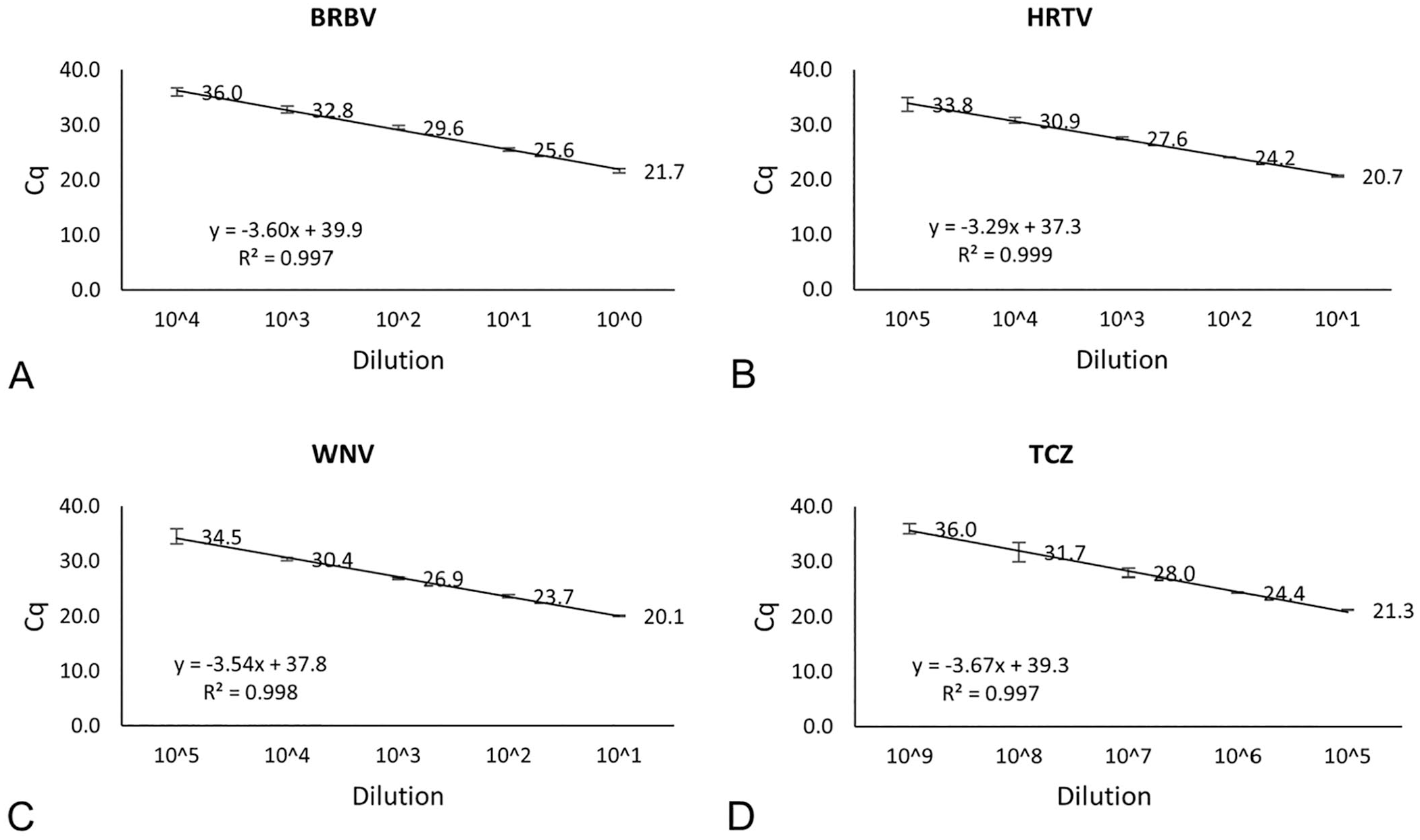
Standard curves of
Performance of RT-qPCR and qPCR on spiked vector samples
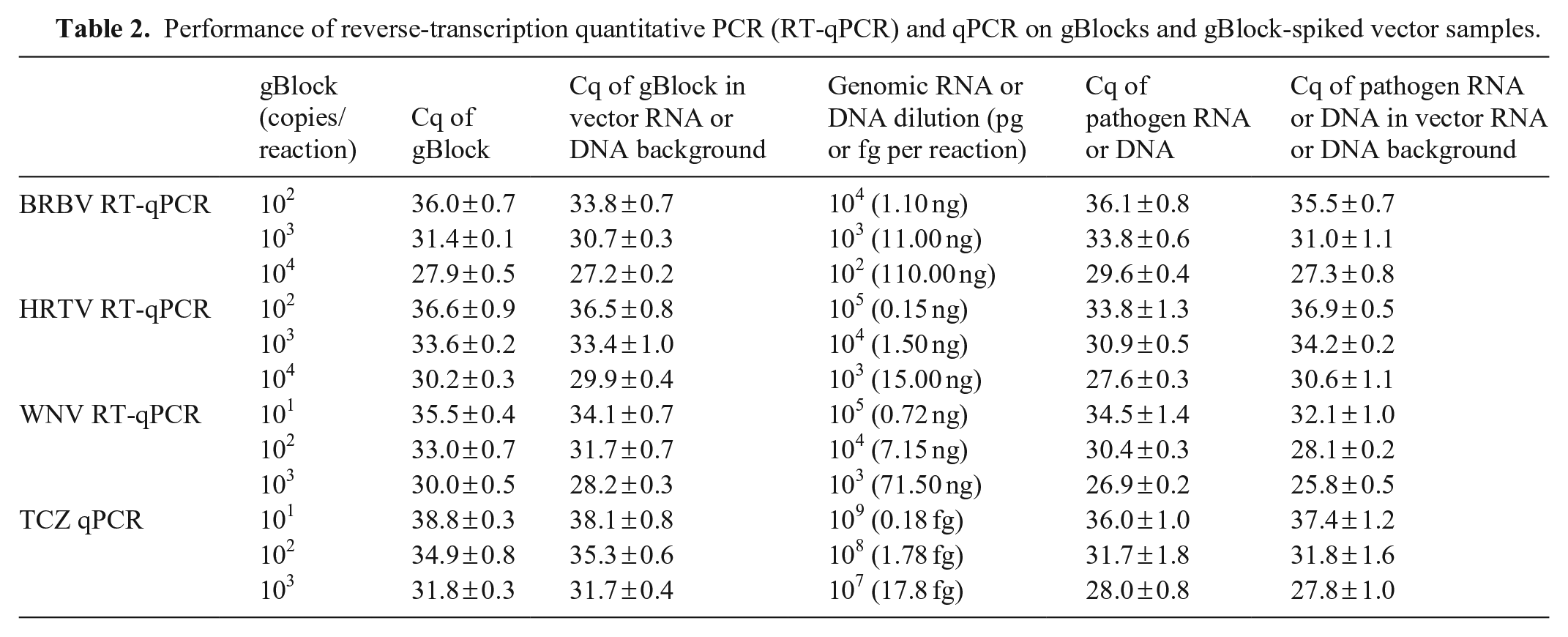
The assays detected all TCZ, BRBV, HRTV, and WNV strains that we tested. When vector RNA and DNA spiked with gBlocks were used as PCR templates, the detection limits for BRBV, HRTV, WNV, and TCZ were 102, 102, 101, and 101 copies per reaction, respectively. The results were essentially the same as those obtained with pure gBlocks. A comparison of Cq values for pure gBlock and gBlock-spiked vector RNA or DNA at 3 concentrations revealed no significant difference between the 2 types of PCR templates at each given concentration (Table 2). When vector RNA and DNA were spiked with gRNA and gDNA, the detection limits of the assays remained at 104 (BRBV), 105 (HRTV), 105 (WNV), and 109 (TCZ) dilutions. There was no significant difference between gRNA or gDNA and vector RNA or DNA spiked with gRNA or gDNA (Table 2).
Performance of reverse-transcription quantitative PCR (RT-qPCR) and qPCR on gBlocks and gBlock-spiked vector samples.
Discussion
Two assays targeting segments 2 and 5 of BRBV 11 have been used in the surveillance of BRBV in field-collected vectors.24–26 However, the detection limit and specificity of these assays have not been evaluated or reported. After assessing the published primers and probes and available BRBV sequences in the NCBI nt database, we decided to design a new set of primers and probe that bind to segment 1. The newly designed primers and probe had a broader coverage than that of the reported primer/probe sets as confirmed by BLAST search. 11 Testing against other vector-borne pathogens verified the specificity of the primers/probe set.
A study reporting 2 sets of primers and probes targeting the S segment of HRTV 22 has been used to detect HRTV in ticks.16,23,26 However, a BLAST search indicated that 1 set of these primers covers 12 of the 13 S segment sequences (100% identify), whereas the other set of primers has 100% match with only 1 sequence deposited in the NCBI nt database. Therefore, it was necessary to design new PCR primers and probe. The newly designed primers and probe sequences matched 100% with all 13 S segment sequences, indicating coverage superior to that of the published primers/probe sets. 22 Testing against other vector-borne pathogens also confirmed the specificity of the HRTV primers/probe set.
Our BRBV and HRTV assays had a detection limit of 102 copies per reaction of gBlocks; detection sensitivities were not affected by vector backgrounds. Because our laboratory routinely extracts RNA from 1 mL of vector homogenate (5 adult ticks or 20–25 nymphs per pool) and uses 1/10 of the resulting RNA in PCR analysis, the detection limit can be converted to 103 copies/mL of tick homogenate. In field-collected tick pools, a surveillance study detected 103.5 pfu/mL of BRBV and 104.0–4.2 pfu/mL of HRTV, respectively.11,22,26 Given that plaque-forming units (pfu) quantify infectious viral particles, the total viral burden (both viable and nonviable) in tick pools could be higher than the pfu counts. Our data collectively indicate that our assays described here are adequate for BRBV or HRTV surveillance.
In contrast to the limited numbers of PCR assays that have been developed for BRBV and HRTV, many WNV PCR assays have been developed.6,7,12,13,33 The primers that we used were published after the emergence of WNV NY99 stain in New York City in the summer of 1999. 12 Over the past 2 decades, WN02 and SW03 have become the dominant strains in the United States. 9 Given that no changes have occurred in the nucleotide sequence of the primer and probe binding regions, the primers were deemed to be adequate. 1 This assay has been used in the detection or surveillance of WNV in people, mosquitoes, birds, or horses.14,30,34 To meet the need for simultaneous testing of multiple pathogens, we modified the assay protocol and validated the assay, which showed excellent sensitivity and specificity. A study using the primers/probe set detected as few as 101.7–2.3 pfu of WNV/mL in infected mosquitoes, C. pipiens quinquefasciatus. 21 The same primers/probe set also showed high sensitivity in our study, with a detection limit of 10 copies per reaction, and the sensitivity was not affected by the vector background. Thus, the modified assay is suitable for detecting WNV in mosquitoes.
A publication summarized the sensitivity, accuracy, and specificity of various TCZ PCR assays. 28 An international study across 16 countries compared 48 PCR assays to detect TCZ in blood samples from Chagas disease patients and identified the 4 best assays. 27 Two of them are conventional PCR, one is a SYBR Green qPCR, and the last one is a TaqMan qPCR 18 that has a detection limit of 2 parasites/mL of blood. Another comparative study, including the 2 conventional PCR assays and the abovementioned TaqMan qPCR assay, suggests that TaqMan qPCR had a higher sensitivity than the conventional PCR assays, with detection limits of 4–40 copies of target region per reaction. 29 The TaqMan PCR assay was also applied to a surveillance study to detect TCZ in triatomine vectors across the United States. 5 We adopted the published qPCR primers and probe (Cruzi 1, 2, 3). We detected a weak cross-reaction with L. major DNA at high concentrations. Given that 1) T. cruzi is transmitted by a blood-sucking triatomine bug, 2) T. vaginalis is a sexually transmitted pathogen, and 3) L. major is transmitted by the sand fly vector, the cross-reaction is not expected to have any impact on the TCZ qPCR assay in terms of pathogen surveillance. In addition, a cross-reaction of this assay has been reported with T. rangeli. 29 However, we performed a BLAST analysis and did not find high similarities between the sequences of TCZ primers and probe and the T. rangeli genome. Regarding the assay sensitivity, our study demonstrated that the TCZ TaqMan qPCR could detect as few as 10 copies of gBlock or 0.003 parasites per reaction, equivalent to 0.6 parasites/mL, which is more sensitive than the published detection limit of 2 parasites/mL. 18 It is noteworthy that the TCZ assay targets the satellite DNA, and different discrete typing units of T. cruzi (dtu TcI–VI) have different numbers of repetitive sequences. Therefore, PCR analytical sensitivity may vary among T. cruzi strains. 19
Because extraction of nucleic acids from vector homogenates was a mature technique, we did not focus on RNA or DNA extraction from infected or spiked vectors. No attempt was made to multiplex these assays because the vectors transmitting these pathogens are different, and the surveillance needs may vary depending on the geographic location. The assays utilize the same and similar thermocycling profile(s), enabling high-throughput testing in laboratories. Given the excellent performance, our assays can be used as standardized methods for rapidly screening vectors for the presence of pathogens by diagnostic and public health laboratories.
Footnotes
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
Our research project “Emerging Vector-Borne Diseases Assay Development” was funded by the Missouri Department of Health and Senior Services.