
Abstract
We used unbiased next-generation sequencing (NGS) to detect unknown viruses in cats. Serum or plasma samples were obtained from clinically ill cats with suspected acute viral infections. Nucleic acid was extracted from serum or plasma samples to construct a complementary DNA library for NGS. Comprehensive nucleotide sequencing analyses enabled detection of the genomes of various viruses, including the severe fever with thrombocytopenia syndrome virus, feline immunodeficiency virus, feline morbillivirus, parvovirus, and Torque teno felis virus. Our findings indicate that comprehensive nucleotide analyses of serum or plasma samples can be used to detect infections with unknown viruses in cats.
Laboratory methods commonly used to test for viral infections are based on pathogen-targeted detection, including antigen–antibody reactions and nucleotide sequence–specific amplification. When a pathogen is unknown, isolation of the virus in cell cultures and experimental infections using animal models are the gold standards for viral identification. However, viral replication can be difficult under experimental conditions, and viral identification can be time-consuming; hence, prompt and concise nontargeted methods would be useful to detect unknown viruses. Comprehensive nucleotide sequence analyses using next-generation sequencing (NGS) provide a powerful new strategy for identifying unknown viruses. These analyses enable researchers to detect the viral genome from the NGS dataset without the need to focus on a specific presumptive virus. Novel viruses have been identified via unbiased NGS from blood samples from humans and horses.3,10 We used unbiased NGS to detect viral genomes in serum and plasma samples from cats with suspected natural acute viral infections.
Blood samples were obtained from 11 household cats admitted to the Kagoshima University Veterinary Teaching Hospital (Kagoshima, Japan) or other veterinary hospitals for diagnosis and treatment. Cases were selected at the discretion of the veterinarian in charge, considering the possibility of severe fever with thrombocytopenia syndrome virus (SFTSV; Phenuiviridae, Bandavirus, Dabie bandavirus) infection. Cats that had spent time outdoors, had a fever of unknown origin, and had leukopenia, thrombocytopenia, or hyperbilirubinemia were preferred for analysis (Table 1). One cat (case 1) was diagnosed with SFTS based on clinical findings and viral genome detection via SFTSV-specific conventional reverse-transcription (RT)-PCR. 15 The other 10 cats had negative RT-PCR and specific antibody test results for SFTSV.
Viral genomes detected with next-generation sequencing and their relative abundances.
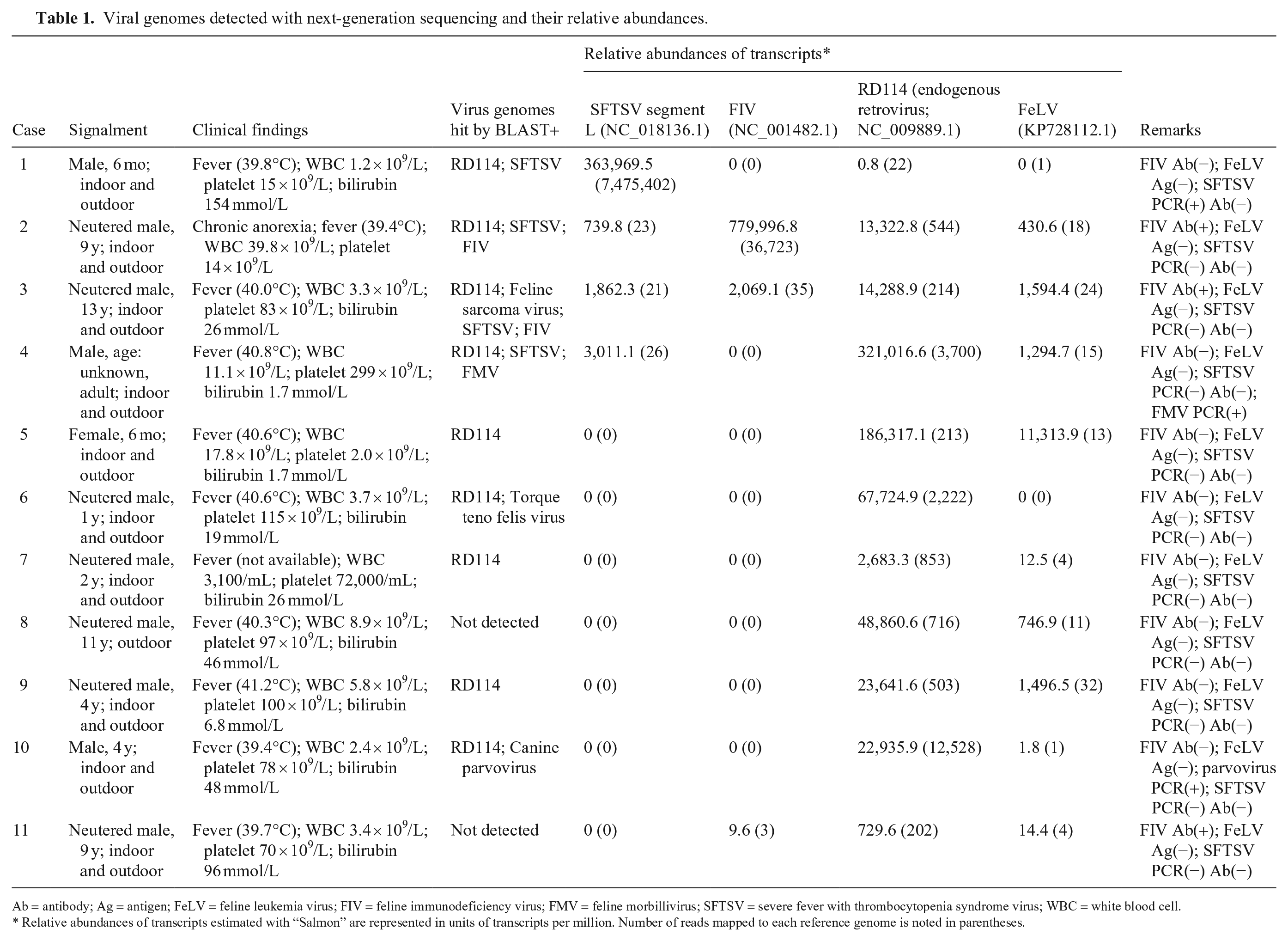
Ab = antibody; Ag = antigen; FeLV = feline leukemia virus; FIV = feline immunodeficiency virus; FMV = feline morbillivirus; SFTSV = severe fever with thrombocytopenia syndrome virus; WBC = white blood cell.
Relative abundances of transcripts estimated with “Salmon” are represented in units of transcripts per million. Number of reads mapped to each reference genome is noted in parentheses.
Cell-free specimens (serum or plasma) were used to investigate the non-host origin genome. Total nucleic acid (DNA and RNA) was extracted from 200 μL of each serum or EDTA-treated plasma sample (magLEAD automatic extraction system, magLEAD Dx SV reagent; Precision System Science). For heparinized plasma samples, RNA was extracted (High Pure viral nucleic acid isolation kit, or High Pure viral RNA isolation kit; Roche) per the manufacturer’s instructions without adding the carrier RNA supplied with the kit. The complementary DNA (cDNA) library was synthesized from the extracted RNA and amplified (Ovation RNA-seq system V2; NuGEN) per the manufacturer’s instructions. The library samples were submitted to Macrogen Japan for NGS analysis. The cDNA library for the NGS was constructed at Macrogen using the TruSeq DNA nano library prep kit (Illumina).
NGS analyses were performed in 2 series, depending on when the blood samples were collected: series 1 (cases 1–4) and series 2 (cases 5–11). For each series of analyses, the individual samples were indexed by ligating barcode adapters to identify the read origin and then mixed for data acquisition in a single lane. The NGS data were obtained using the NovaSeq 6000 system (Illumina); ~ 29–37 million 150-bp paired-end reads were obtained per sample. The obtained reads were trimmed and filtered with the phred quality score. 5 The trimmed sequences were then analyzed with the Galaxy web-based genomics analysis platform (https://usegalaxy.org) and mapped to the feline genome (Felis_catus_9.0: GCA_000181335.4) using “HISAT2”. 12 Unmapped reads were assembled de novo using the Trinity program. 9 A homology search of the assembled contigs (minimum contig length: 200 bp) was conducted against viral reference genomes BLAST + nblast (https://blast.ncbi.nlm.nih.gov/Blast.cgi). 1 The viral sequences hit by BLAST were filtered under the conditions of alignment length > 200 bp and percentage of identical nucleotide matches in the region > 80%. Nonvertebrate host viruses and contigs highly homologous to the feline genome were excluded. If a contig hit multiple viral sequences, the most homologous virus with the lowest expectation value was selected. The unmapped reads on the feline genome via the “HISAT2” program were mapped again on the viral sequences hit by BLAST using the “Salmon” quantification program, 16 which allows quantification of transcripts and counting mapped reads. Relative abundances of reads mapped on the viral sequence are represented in units of transcripts per million (Table 1).
One cat diagnosed with SFTS (case 1) had a large number of SFTSV-related reads. A few SFTSV-related reads were detected in other cats analyzed in the same lane (cases 2–4), but these cats tested negative on the SFTSV-specific RT-PCR and for anti-SFTSV IgG and IgM antibodies. 15 Feline immunodeficiency virus (FIV) was hit by BLAST in 2 of 3 FIV antibody–positive cats (Table 1). Circulating FIV RNA loads in plasma are reported to increase as the clinical FIV infection stage progresses from asymptomatic to immunodeficient and have been reported to range from < 1 × 103 to 2 × 108 copies/mL. 8 Case 2 had a high number of FIV-related reads. This cat had a fever of unknown origin lasting several months. This clinical sign was consistent with the immunocompromised condition observed in the terminal stage of FIV infection. BLAST did not hit FIV in one FIV antibody–positive cat (case 11). Although the FIV-related reads were present as per the “Salmon” results, the viral load was so low that the reads were not assembled to FIV-related contigs meeting the cutoff value for BLAST (length > 200 bp) in this case.
Feline morbillivirus (FMV) is thought to be associated with chronic renal disease in cats. Previous reports have indicated that the FMV viral genome is occasionally detected from urine samples but rarely detected from blood samples when using RT-PCR.4,7 BLAST detected the viral genome in blood samples from one cat (case 4), and the viral genome was confirmed via conventional RT-nested PCR conducted as per a previous report. 6 This cat had clinical signs consistent with acute infection at the time of sample collection, and clinically recovered within 2 wk.
We detected the parvovirus genome in one cat (case 10), and the result was confirmed via TaqMan real-time PCR as per a method described previously. 21 This cat had clinical signs consistent with acute parvoviral infection, including fever and vomiting, but no diarrhea was observed at the time of sample collection.
We detected the Torque teno felis virus in case 6. Pathogenicity of this virus is unknown; the genome has been reported in 10–35% of feline samples obtained at animal hospitals.11,23 We had designed our study to detect RNA viruses primarily. Detection of DNA viruses (parvovirus and the Torque teno felis virus) may have been related to the procedure used, in which both DNA and RNA were extracted, and the DNA was not digested before cDNA synthesis. Although covering both DNA and RNA viruses requires larger amounts of sequencing data, NGS data can be acquired in an analysis when determining unknown viral infections, which may be more convenient and cost-effective in one sequencing run.
Identification of the feline leukemia virus (FeLV) and other type C retroviruses was difficult in cats in our study. Feline genomes contain dozens of endogenous retroviral genomes similar to those of FeLV.2,17,19,20 These genomes are transcribed and produce both noninfectious viruses and the infectious virus, RD114, which is produced by genome recombination. 18 The consistent detection of retrovirus-related reads in our study might reflect that these virus-like endogenous genomes were circulating in the serum. Identification of the origin of the erroneous short reads produced via NGS may be impossible.
We demonstrated that unbiased NGS-based analysis of nucleic acids from sera or plasma is a potentially powerful tool for detecting unknown viruses in cats. However, our study indicated that these analyses involve a risk of data contamination. With NGS, multiple samples are commonly analyzed in a single lane by adding index adapters to reduce the cost per sample. Multiplexing can induce contamination of the read data between samples by index misassignment, known as index hopping. A few SFTSV-related reads were detected in 3 SFTSV PCR–negative samples (cases 2–4) that were analyzed in the same lane as the SFTSV-infected cat (case 1). Although low-level read misassignment may be permissible for some research objectives (e.g., transcriptome analyses), data contamination should be prevented when applying NGS to detect infections. Techniques and methods that reduce the level of index hopping include using dual-indexed libraries. 13 However, it can be difficult to reduce the frequency of misassignment to a negligible level for infectious microorganism detection when applying multiplexing. Contamination of lab reagents can also potentially contaminate data. Setting a cutoff value for viral detection may increase the specificity but reduce the sensitivity, as shown in our FIV antibody–positive case (case 11).
NGS results should be interpreted carefully, and the results should be confirmed by target detection, such as by conventional PCR. We demonstrated that the viral genome could be identified via unbiased NGS-based analysis directly from feline serum or plasma samples. Convenient bioinformatic tools and analysis pipelines have been developed to detect pathogens.14,22 Progress in analytical procedures as well as decreased costs will enable unbiased NGS-based analysis to become a common laboratory procedure for detection of unidentified pathogens in clinical veterinary medicine.
Footnotes
Acknowledgements
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
This work was supported by a Japan Society for the Promotion of Science Grant-in-Aid for Scientific Research (KAKENHI; grant JP16K08057).