
Abstract
Equine infectious disease outbreaks may have profound economic impact, resulting in losses of millions of dollars of revenue as a result of horse loss, quarantine, and cancelled events. Early and accurate diagnosis is essential to limit the spread of infectious diseases. However, laboratory detection of infectious agents, especially the simultaneous detection of multiple agents, can be challenging to the clinician and diagnostic laboratory. Next-generation sequencing (NGS), which allows millions of DNA templates to be sequenced simultaneously in a single reaction, is an ideal technology for comprehensive testing. We conducted a proof-of-concept study of targeted NGS to detect 62 common equine bacterial, viral, and parasitic pathogens in clinical samples. We designed 264 primers and constructed a bioinformatics tool for the detection of targeted pathogens. The designed primers were able to specifically detect the intended pathogens. Results of testing 27 clinical samples with our targeted NGS assay compared with results of routine tests (assessed as a group) yielded positive percent agreement of 81% and negative percent agreement of 83%, overall agreement of 81%, and kappa of 0.56 (moderate agreement). This moderate agreement was likely the result of low sensitivity of some primers. However, our NGS assay successfully detected multiple pathogens in the clinical samples, including some pathogens missed by routine techniques.
Introduction
The laboratory detection of infectious disease agents, especially in the case of mixed infections with 2 or more pathogens, is challenging to the clinician and the diagnostic laboratory. Routinely, submission is required of multiple samples to separate laboratory sections. Clinical microbiology has traditionally relied on isolation of pathogens by culture, followed by biochemical and other tests to identify the genus or species. Also, molecular testing, including PCR, has been used widely for detection of many viral and bacterial pathogens.7,10,23 However, because most of these tests detect only one or a few pathogens, multiple tests are often required to identify the potential causative agents, especially when clinical signs are nonspecific. Next-generation sequencing (NGS) can overcome this limitation by simultaneously testing for numerous infectious agents in a single tube. Therefore, NGS-based testing is being adopted in diagnostic microbiology.3,9,21
NGS studies of microorganisms typically follow 1 of 2 strategies: whole-genome sequencing (WGS) or targeted sequencing.24,28 WGS has the potential to sequence all nucleic acids within a sample and provides complete characterization of the genomic content. However, this unbiased sequencing requires a substantial amount of sequence depth, particularly for viruses, to separate low-prevalence pathogens from the overwhelming contribution of host nucleic acid. 17 In contrast, targeted sequencing uses target-specific primers for PCR-mediated amplification, such that the genomic regions of interest are enriched and selectively sequenced. Compared to unbiased sequencing, targeted sequencing provides better coverage, specificity, and ease of downstream analysis.2,12,14,20 Hence, targeted NGS is a promising tool to provide comprehensive assays for the detection of known, clinically relevant pathogens from a variety of specimens, particularly for cases with nonspecific disease indications that may be associated with multiple infectious agents. Studies have demonstrated the feasibility of using targeted sequencing for clinical infectious disease testing.1,15,30 Further, combining targeted sequencing with WGS can significantly improve the recovery of whole genome sequences of viruses (e.g., Hantaan orthohantavirus 18 ) directly from clinical samples.
We evaluated the use of targeted NGS for detection of 62 equine pathogens in clinical samples. We tested 27 clinical equine samples with the targeted NGS assay as well as with routine laboratory methods to evaluate the feasibility of applying a targeted NGS technique in a clinical molecular laboratory for both testing of clinical submissions and disease surveillance.
Materials and methods
Design of amplicon panel primers
Primers were designed for 62 equine pathogens selected for their relevance to clinical equine disease observed in the field, including bacterial, fungal, viral, and parasitic pathogens (Suppl. Table 1). The primers were designed to target specific regions of each pathogen (~200 bases per target region) based on suitable regions published in the literature, using the AmpliSeq Designer (Ion Torrent; Thermo Fisher). Based on in silico analysis of primer specificity using NCBI BLAST (https://blast.ncbi.nlm.nih.gov/Blast.cgi), changes were made to the design with the assistance of the AgriSeq bioinformatics team (Ion Torrent; Thermo Fisher). Because variation in targeted organisms could cause individual primers to fail, multiple primers per targeted organisms were designed to provide redundancy. Based on the assay design, the primers were separated into 2 primer pools to reduce binding between the primers.
Nucleic acid extraction, library preparation, and NGS
Total nucleic acid (both DNA and RNA) was isolated (DNeasy blood and tissue kit; Qiagen). A modification of the animal tissue protocol was employed as described previously. 1
Automated library preparation, template preparation, and chip loading were performed (Ion Chef instrument; Thermo Fisher) as described elsewhere. 1 Briefly, reverse-transcription PCR (RT-PCR) using the designed primer pools and library preparation were performed (Ion Chef, AmpliSeq kit for Chef DL8; Thermo Fisher), according to the manufacturer’s protocol. This kit allowed the preparation of 8 barcoded Ion AmpliSeq libraries per Ion Chef run (8 different clinical cases). Then, 50 pmol of the 8 mixed libraries were used to prepare the template and load a chip (Ion 314 chip, Ion Chef instrument, Ion PGM kit; Thermo Fisher), according to the manufacturer’s instructions. Finally, the libraries were sequenced (Ion PGM Hi-Q View sequencing kit, Ion Torrent personal genome machine; Thermo Fisher), according to the manufacturer’s instructions.
Automated preparation minimizes sample handling and lowers chance of contamination as well as provides reproducible chip loading. This automated workflow generated results within 2–3 d.
Data analysis
A reference file containing the sequences of the targeted pathogens obtained from GenBank was constructed and uploaded to the Ion Torrent suite software (Thermo Fisher). These files were used for initial data analysis with the Torrent suite software, including read trimming, assembly with SPAdes (http://coolgenes.cahe.wsu.edu/ion-docs/Assembler-SPAdes-Plugin.html), and mapping to the reference file. Then, the Bam files were downloaded and evaluated with Geneious software (v.9.1.2; Biomatters). Finally, pathogen identifications were confirmed with BLAST. BLAST (E) values <1 were considered acceptable. The lower the E-value, or the closer it is to zero, the more “significant” the match of the alignments (https://blast.ncbi.nlm.nih.gov/Blast.cgi?CMD=Web&PAGE_TYPE=BlastDocs&DOC_TYPE=FAQ#expect).
Assay analytical performance
Both analytical sensitivity (ASe; relative limit of detection [RLOD]) and analytical specificity (ASp) were determined to assess the analytical performance of the assay. The relative ASe of a subset of the organisms was assessed for proof-of-concept testing of the design for detection of these pathogens in clinical samples. This was performed by testing relatively known quantities (based on quantitative PCR [qPCR] or reverse-transcription real-time PCR (RT-rtPCR) results, cycle threshold [Ct] values) of DNA from representatives of the viral, bacterial, fungal, and parasitic pathogen groups, as well as viral RNA. Seven clinical samples containing very low levels (Ct 30–35) of organisms, including Clostridioides difficile, Clostridium perfringens, Salmonella spp., Streptococcus equi subsp. equi, Neorickettsia risticii, equine herpesvirus 1 (EHV-1), and West Nile virus (WNV), were tested to determine the RLOD. Typically, for qPCR or RT-rtPCR testing, higher Ct values (>35) are considered suspect because they may only represent fluorescence artifacts or cross-contamination. 6 The qPCR or RT-rtPCR assays were performed at the Tifton Veterinary Diagnostic and Investigational Laboratory, College of Veterinary Medicine, University of Georgia (TVDIL; Tifton, GA), Indiana Animal Disease Diagnostic laboratory, College of Veterinary Medicine, University of Purdue (ADDL; West Lafayette, IN), and Veterinary Diagnostic Laboratory, University of Kentucky (UKVDL; Lexington, KY) with laboratory-validated procedures. These laboratories are accredited by the American Association of Veterinary Laboratory Diagnosticians (requirements are based upon the ISO/IEC 17025:2005 standard, https://www.iso.org/standard/39883.html).
ASp was performed to determine the ability of the assay to detect the targeted pathogens without being affected by specimen-related conditions or cross-reactivity and/or interference of the host nucleic acid. To assess the detection ability of the assay and to better mimic clinical samples, a sample known to contain 1 pathogen based on previous testing was spiked with equal amounts of 4 other pathogens. The assay was evaluated by testing validated isolates or reference strains of most of the pathogens (bacteria, parasites, fungi, and viruses) that can be detected using this panel (Table 1) Eight pathogens were not tested, namely Clostridium tetani, Taylorella equigenitalis, Babesia caballi, Theileria equi, Leishmania sp., Neospora hughesi, Trypanosoma cruzi, Halicephalobus sp., Campylobacter coli, Venezuelan equine encephalitis virus (VEEV), equine coronavirus (ECoV), and equine adenovirus 1 and 2.
List and source of isolates and reference strains of the pathogens that can be detected by the targeted NGS assay.

A = USDA National Veterinary Services Laboratories; B = detected in a diagnostic sample and identified and isolated in the TVDIL according to a validated protocol as follows: bacterial isolates were identified via biochemical testing and a commercial microtiter system; viral isolates were identified via fluorescent antibody test or RT-qPCR; C = E. coli Reference Center, Pennsylvania State College of Agriculture Sciences, University Park, PA; D = detected in a diagnostic sample and identified via specific qPCR by the Clinical Immunology and Virology laboratory, College of Veterinary Medicine, University of Tennessee (Knoxville, TN); E = detected in a diagnostic sample by specific qPCR by the Indiana Animal Disease Diagnostic laboratory, Purdue College of Veterinary Medicine, University of Purdue (West Lafayette, IN); F = validated reference kindly provided by Drs. Chunlei Su and Rick Gerhold, University of Tennessee; G = validated reference kindly provided by Dr. Daniel K. Howe, University of Kentucky (Lexington, KY); H = validated reference kindly provided by Dr. Amy Swinford, Texas A&M Veterinary Medical Diagnostic Laboratory (College Station, TX).
Assay clinical evaluation and statistical analysis
Determination of the diagnostic sensitivity and specificity of an assay requires the evaluation of every detected pathogen in every clinical case and comparing the sensitivity and specificity with that of gold standard test results for known positive and negative samples. Instead, we compared the NGS panel with routine laboratory methods for detection of these pathogens in clinical samples for proof-of-concept testing, considering that the purpose of the NGS test would be to replace these routine methods. We determined the positive percent agreement (PPA), negative percent agreement (NPA), and total agreement between the NGS method and the routine methods used collectively for case diagnosis. We included in the comparative study 27 equine clinical cases that were submitted to TVDIL, ADDL, UKVDL, and Pennsylvania Animal Diagnostic Laboratory System–New Bolton Center, School of Veterinary Medicine, University of Pennsylvania (PADLS-NBC; Kennett Square, PA) in 2018–2019. We evaluated a broad range of sample types and clinical conditions for our proof-of-concept testing. Samples had been sent to each laboratory for diagnostic purposes, and not all samples were submitted with complete histories.
Each case was tested via the designed targeted NGS assay as well as with routine laboratory methods. The routine tests included bacterial culture, ELISA, PCR, RT-rtPCR, and qPCR. Each of the routine tests had been done by the submitter laboratory following their laboratory-validated procedures. The relative PPA and NPA were assessed with respect to routine methods as a group. 11 Also, the overall agreement and Cohen kappa were assessed by comparing the new assay to routine methods as a group (if any of a group of agents was not detected by the new assay or routine methods, the result for the test group was considered negative). Cohen kappa is the standard agreement coefficient that considers the possibility of the agreement occurring by chance. Cohen kappa is always ≤1; a value of 1 indicates perfect agreement between 2 tests, and 0 indicates that any agreement is the result of chance. 16
Results
Our newly designed primer pools were able to specifically detect and/or sequence the target region of all bacteria, fungi, parasites, and viruses that we tested. For the LOD, 7 representative bacteria, viral DNA, and viral RNA were tested. The targeted equine NGS panel was able to detect pathogens with Ct values of 30–35 for 5 of the tested targets. Primer sets that targeted N. risticii and C. difficile resulted in poor sequencing coverage (low number of reads) or no sequence, respectively, when tested with clinical samples that were positive by qPCR (Ct = 29.5).
We evaluated the targeted NGS method using 27 equine clinical samples representing neurologic (n = 2), abortion (n = 3), intestinal (n = 12), and respiratory (n = 9) diseases, and a cutaneous wound (n = 1), all tested previously using other routine methods. Targeted NGS not only successfully identified multiple pathogens in the clinical samples in concordance with routine methods but also identified pathogens not detected by the routine techniques (Table 2). Specifically, targeted NGS detected EHV-1 in 1 of 2 neurologic and in 3 of 3 abortion cases that were positive via qPCR with Ct values of 15–32.9. In the second neurologic case, WNV was detected by both targeted NGS as well as RT-rtPCR, with good correlation with pathology findings. Of 12 intestinal samples, there was agreement between targeted NGS and routine microbiologic techniques in 8 of 12 cases, with good correlation with pathology findings in 6 of 11 samples for which pathology results were available. Cases in which targeted NGS and routine microbiologic results were not correlated included one fecal sample for which targeted NGS failed to detect any pathogens but in which rotavirus, C. difficile, and C. perfringens were detected by RT-rtPCR and qPCR with Ct values of 37.7, 37.1, and 37.1, respectively. Interestingly, pathology findings included mycotic pneumonia and cecocolonic hyperemia without evidence of overt enterocolitis, making interpretation of the clinical significance of these PCR findings difficult. The other intestinal cases without agreement between microbiologic testing methods and NGS included 2 cases in which pathogens in intestinal contents were only identified using targeted NGS. In one case, Actinobacillus sp. was identified in intestinal contents from a horse with enteritis confirmed by pathology; in another case, Pseudomonas aeruginosa septicemia was diagnosed by splenic culture and later confirmed by pathology.
Clinical case comparative study results of the targeted NGS assay and the routine methods.
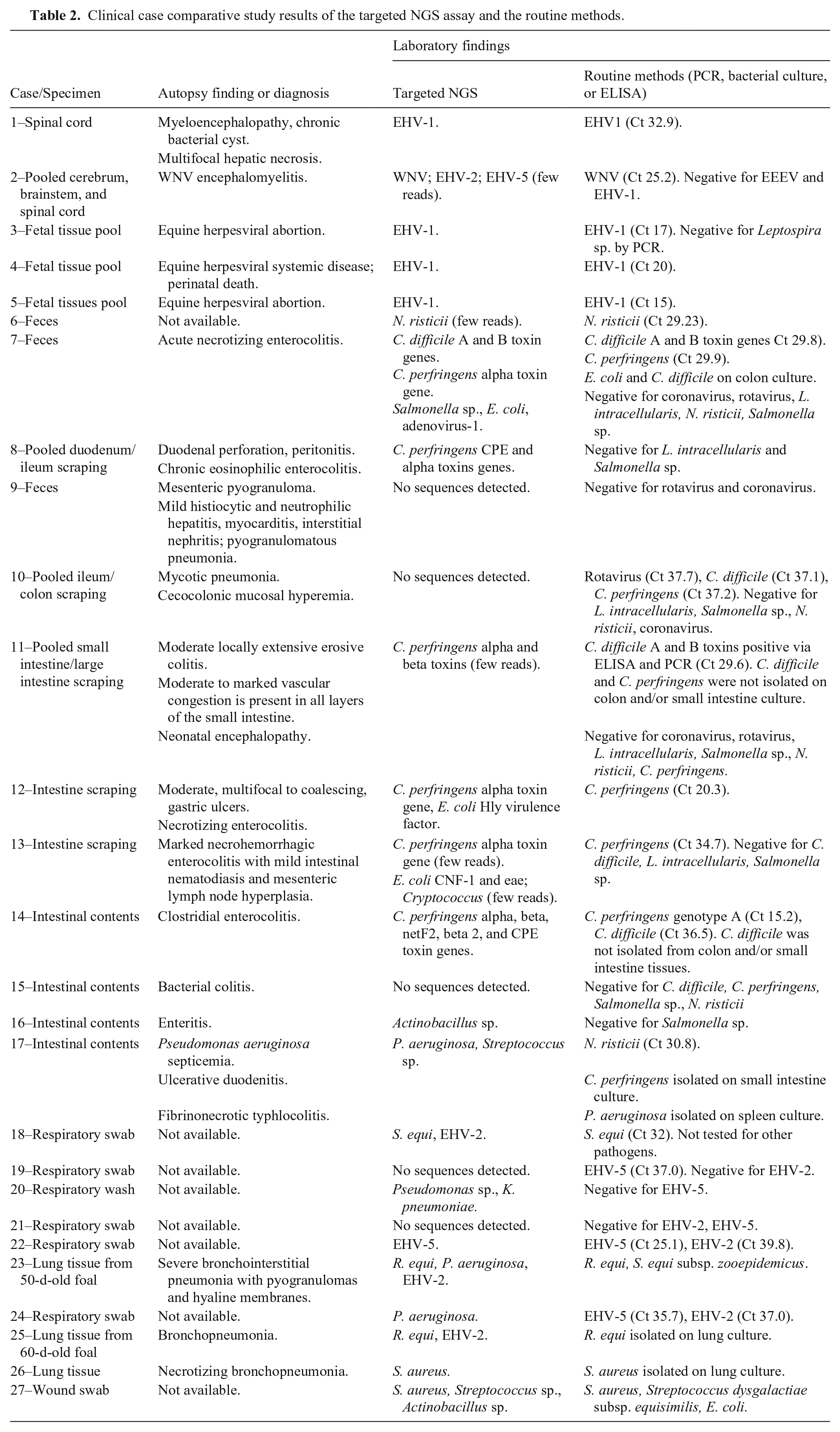
Among the 9 samples from respiratory cases, comprising swabs, fluid from transtracheal washes, and lung tissue, only 3 of 9 had pathology results available for comparison, but they had good correlation between lesions and pathogens identified by both testing methods. However, our targeted NGS failed to amplify EHV-5 in 1 case (Ct 37.0) without available pathology results, and failed to amplify EHV-2 in 2 cases (Ct 37.0 and 39.8, respectively); in 1 of these 2 cases, EHV-5 was detected by both methods. In one case of a wound, there was good correlation between targeted NGS and routine bacterial culture.
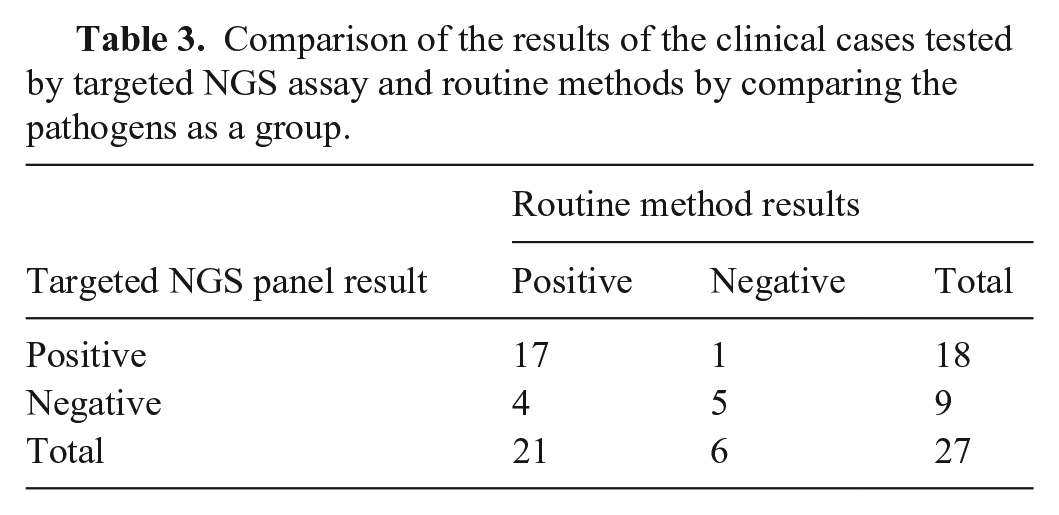
Based on our comparative study, the overall agreement between our new assay and the routine methods used was 81%, with κ = 0.56 (moderate agreement). The PPA and NPA of the targeted NGS were 81% and 83%, respectively (Table 3).
Comparison of the results of the clinical cases tested by targeted NGS assay and routine methods by comparing the pathogens as a group.
Discussion
We evaluated the use of targeted NGS to detect equine pathogens directly from clinical samples. Our new assay was able to detect pathogens (Clostridium perfringens, Salmonella spp., Streptococcus equi subsp. equi, EHV-1, and WNV) that had high Ct values (29–32.9) when tested by qPCR, demonstrating the ability of the assay to detect some pathogens even at low levels. Obtaining good sequence recovery directly from clinical samples, particularly in samples with low levels of organism, has been a challenge for the application of NGS technology as a detection tool. However, the use of targeted NGS to generate a large number of sequences for the target gene or region significantly improves the detection of targets in degraded and low-titer samples.1,8,29 Additionally, despite the limited testing performed, we were able to successfully detect pathogens with the targeted NGS method from multiple sample types.
Evaluation of ASe is usually done by testing the LOD with plasmids or in vitro transcribed RNA per target. Considering the lack of availability of all needed reference material to perform this type of testing and the prohibitive cost, we decided to limit this evaluation to RLOD for group representatives. These samples represent the different types of pathogens targeted by the protocol (bacterial DNA, viral DNA, and viral RNA) and were known from previous testing to contain very low levels of organisms (qPCR and RT-rtPCR Ct of 30–35). Although this may not be the recommended way to evaluate the LOD of an assay, it provided an estimate of the RLOD for the NGS design. Our targeted NGS equine assay is intended to detect organisms associated with clinical disease, therefore determining the absolute ASe was considered less important than if this test was intended to test for carrier status or for freedom from disease, for which a very low LOD is needed. Further evaluation of the LOD will be needed before the use of our assay as a routine test.
Not all pathogens for which primers were included in our panel were tested. Included in this group were the foreign animal disease pathogens. Primers for these pathogens were included in our assay to develop a comprehensive test but also as a means of providing expanded surveillance, which could be done along with routine testing with this type of assay. A justification for including some of these primers is the fact that climate change is allowing expansion of pathogens into new areas. It is important to note, however, that if a NGS assay detected a foreign animal disease pathogen, the proper authorities would need to be notified and appropriate confirmatory testing performed.
The overall agreement between the new assay and the routine methods was 81%. The main contributor to the reduced agreement was failure of the NGS method to detect very low levels of some organisms, which were detectable in the samples by qPCR. Primers for organisms that had reduced sensitivity based on this testing were those for N. risticii, C. difficile, EHV-2, and EHV-5. Samples with low amounts of these organisms had poor sequencing coverage (low number of reads) or no sequence.
For respiratory cases evaluated in our study, the significance of the qPCR results with Ct values in the high 30s for EHV-2 and EHV-5 in upper respiratory tract swabs is unknown. Both EHV-2 and EHV-5 produce persistent infections in the host. EHV-5 can be detected in the lungs of unaffected horses, but it has been associated with equine multinodular pulmonary fibrosis.4,19 Similarly, the role of EHV-2 as a pathogen is controversial; some studies have demonstrated its association with upper respiratory tract disease, lymphadenopathy, immunosuppression, and keratoconjunctivitis.5,13,22 Unfortunately, relatively few respiratory cases used in our study had pathology results available (either cytologic or histologic), making interpretation of the findings difficult. Interpretation of any molecular test result, including qPCR and/or NGS, should take into consideration the clinical and pathologic findings (if available) for accurate diagnosis. However, producing an assay with the ability to detect the full range of pathogen shedding, including small amounts that would be expected in convalescent cases, is desirable. At least for some pathogens that we tested, qPCR performed better for detection of very low levels of pathogen.
Most enteric cases had good correlation between microbiologic testing techniques, targeted NGS results, and pathologic findings. However, in 2 enteric cases, in addition to the C. perfringens toxins that were detected by both the new assay and qPCR, our targeted NGS detected some additional pathogens. These included E. coli hemolysin (Hly) virulence factor in intestinal scrapings from a horse with gastric ulcers, and E. coli cytotoxic necrotizing factor 1 (CNF-1) and intimin (eae) virulence factors in intestinal scrapings from a horse with necrohemorrhagic enterocolitis, which also had concurrent C. perfringens alpha toxin and Cryptococcus sp. detected by targeted NGS. Although C. perfringens was detected by qPCR (Ct 34.7), the reason the other pathogens were missed by routine methods is because the specific test needed was not performed or not requested on sample submission. Although the multiple pathogens identified by targeted NGS may represent mixed infection with multiple virulent organisms contributing to clinical disease, additional samples correlating pathologic findings with the presence of virulent pathogens are needed before clinical interpretations can be made.
As demonstrated with our targeted NGS results, an advantage of this method is its ability to not only detect conserved regions of these enteric pathogens but to identify E. coli and C. perfringens virulence factors or toxins to distinguish commensals from pathogenic bacteria. Other potential advantageous applications of NGS methods are to identify genotypic markers of drug resistance and virulence, as well as strain typing. Therefore, targeted NGS could distinguish between wild-type and vaccine strains as well as predict phenotypic antimicrobial resistance by targeting known genetic determinants of antimicrobial resistance.25-27 Detection and characterization of the pathogen from the clinical sample could be done in a single assay.
Overall, our targeted NGS testing method performed favorably, and our pilot study has demonstrated the feasibility of using targeted NGS for equine infectious disease testing. However, redesign of the primer pools for targets with reduced sensitivity as well as further evaluation are recommended to improve the assay.
Supplemental Material
sj-pdf-1-jvd-10.1177_1040638720978381 – Supplemental material for Evaluation of targeted next-generation sequencing for detection of equine pathogens in clinical samples
Supplemental material, sj-pdf-1-jvd-10.1177_1040638720978381 for Evaluation of targeted next-generation sequencing for detection of equine pathogens in clinical samples by Eman Anis, Marcia R. S. Ilha, Julie B. Engiles and Rebecca P. Wilkes in Journal of Veterinary Diagnostic Investigation
Footnotes
Acknowledgements
We thank Dr. Erdal Erol (University of Kentucky, Veterinary Diagnostic Laboratory), Dr. Hemant Naikare (Tifton Veterinary Diagnostic and Investigational Laboratory, University of Georgia), and Dr. Daniel K. Howe (University of Kentucky) for their technical support and help in providing isolates and clinical samples.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received financial support for their research from the Advancement in Equine Research Award from Boehringer Ingelheim. Boehringer Ingelheim had no influence on the study design or interpretation of results.
Supplementary material
Supplementary material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.