
Abstract
The genomic surveillance of porcine reproductive and respiratory syndrome virus (PRRSV) is based on sequencing of the ORF5 gene of the virus, which covers only 4% of the entire viral genome. It is expected that PRRSV whole-genome sequencing (WGS) will improve PRRSV genomic data and allow better understanding of clinical discrepancies observed in the field when using ORF5 sequencing. Our main objective was to implement an efficient method for WGS of PRRSV from clinical samples. The viral genome was purified using a poly(A)-tail viral genome purification method and sequenced using Illumina technology. We tested 149 PRRSV-positive samples: 80 sera, 33 lungs, 33 pools of tissues, 2 oral fluids, and 1 processing fluid (i.e., castration liquid). Overall, WGS of 67.1% of PRRSV-positive cases was successful. The viral load, in particular for tissues, had a major impact on the PRRSV WGS success rate. Serum was the most efficient type of sample to conduct PRRSV WGS poly(A)-tail assays, with a success rate of 76.3%, and this result can be explained by improved sequencing reads dispersion matching throughout the entire viral genome. WGS was unsuccessful for all pools of tissue and lung samples with Cq values > 26.5, whereas it could still be successful with sera at Cq ≤ 34.1. Evaluation of results of highly qualified personnel confirmed that laboratory skills could affect PRRSV WGS efficiency. Oral fluid samples seem very promising and merit further investigation because, with only 2 samples of low viral load (Cq = 28.8, 32.8), PRRSV WGS was successful.
Keywords
Introduction
Porcine reproductive and respiratory syndrome (PRRS) is an important viral disease causing severe economic losses in North America, with estimated losses of $663 million and $150 million per year in the United States and Canada, respectively.19,35 Therefore, considerable efforts and resources are allocated to attempt to control and contain the virus. Among those efforts, biosecurity measures, vaccination, and epidemiologic surveillance are the most prominent.29,37 The disease has many clinical manifestations, but the 2 most common are reproductive disorders in sows and gilts (characterized by late abortions with an increased number of stillbirths and mummified fetuses, including weak-born piglets)4,7,21,30,41 and respiratory problems in pigs of all ages (characterized by interstitial pneumonia).1,4,8,15,41
PRRS virus (PRRSV; Nidovirales, Arteriviridae, Betaarterivirus suid), the etiologic agent of PRRS,22,34 is an enveloped virus of 50–65 nm diameter. 44 The PRRSV genome is composed of a positive single-stranded RNA of ~ 15 kb nucleotides (nt), and it encodes at least 11 open reading frames (ORFs). 32 Approximately three-quarters of the genome is composed of ORF1 at the 5′-end of the genome, which encodes for nonstructural proteins; the ORFs located at the 3′-end (ORF2–7) encode mostly for virion structural proteins.10,20,34 Moreover, its viral genome extremities are capped at the 5′-end, and with a polyadenylated tail (poly(A)-tail) at the 3′-end.10,20 The pathogenicity of PRRSV is multigenic, whereas several lineages of PRRSV exist within both PRRSV species (i.e. Betaarterivirus suid 1 and 2, commonly named PRRSV types 1 and 2 or PRRSV-1 and -2, respectively) and, consequently, the virulence of the strains is variable (from low to high). 34 Molecular epidemiologic tools have been developed to help in the control of PRRSV and to understand the links between a specific PRRSV strain with: (1) the origin of an outbreak, and (2) its genetic proximity with vaccine strains, to estimate their potential efficacy.25–27
The ORF5 gene of PRRSV has been selected for the epidemiologic surveillance of PRRSV strains because: (1) it is hypervariable,27,40 and (2) it encodes a protein (GP5) that induces the synthesis of neutralizing antibodies in pigs.13,39 This GP5 antigenicity property has contributed to the selection of ORF5 for PRRSV surveillance despite the fact that other viral proteins are involved in the neutralizing antibody recognition of the virion. 6 Most reported studies have used ORF5 for the classification of PRRSV. 27 Moreover, ORF5 sequencing is part of the strategy that has been put in place to control PRRSV.25,26 As an example, with the agreement of all of the swine veterinarians in the province of Québec (Canada), the Molecular diagnostic laboratory (MDL) of the Service de diagnostic, Faculté de médecine vétérinaire (FMV), Université de Montréal (UdeM), is the depository of a databank containing almost all PRRSV ORF5 nt sequences that have been sequenced since ~ 2010. This agreement has set up the rules for data sharing between veterinarians. To date, at least 4,695 PRRSV ORF5 nt sequences are included in this databank. Noteworthy, some researchers have recently reported that 11% of the PRRSV strains were misclassified by the ORF5 sequencing method (i.e., PCR amplification of the ORF5 gene followed by the sequencing of ORF5 PCR amplicons by the Sanger-based sequencing method) compared to PRRSV whole-genome sequencing (WGS), because of viral genome recombination events and simultaneous coinfection of 2 significantly different PRRSV strains in some swine clinical samples. 24 Thus, the ORF5 genomic methodology that is currently used can cause classification errors and therefore mislead veterinary interventions.
Over the past few years, next-generation sequencing (NGS) has been a very powerful tool for the detection and discovery of viruses in a large diversity of environments, sample types, and hosts.9,12,14,18 In fact, NGS has allowed the discovery and sequencing of the entire viral genome of new swine viruses, such as the atypical porcine pestivirus and porcine circovirus 3.16,38 Therefore, there is an urge to improve the present ORF5 molecular epidemiologic tool for the surveillance of PRRSV through the use of NGS tools. During the last North American PRRS Symposium and Conference of Research Workers in Animal Diseases (CRWAD) joint meeting (Nov 2019, Chicago, IL: https://vetmed.illinois.edu/vet-resources/continuing-education/north-american-prrs-symposium/, https://crwad.org/crwad2019/), several speakers reported the WGS of PRRSV using different technologies, such as MiSeq (Illumina) and MinION (Oxford Nanopore Technologies). Unfortunately, a lack of data was obvious regarding the efficacy of those WGS methods with various swine clinical samples.
Therefore, we implemented an efficient method for WGS of PRRSV from different types of swine clinical samples that are routinely submitted for diagnostic and surveillance purposes, allowing its use in a real veterinary diagnostic laboratory environment.
Materials and methods
Swine clinical samples
Convenience swine clinical samples that tested positive for PRRSV by a reverse-transcription quantitative PCR (RT-qPCR) assay were selected for WGS with the goal of obtaining the entire PRRSV viral genome. The clinical samples were submitted between December 2015 and July 2019; most were collected in 2017 (n = 43) and 2018 (n = 46). The samples were mainly tested by the MDL using a PRRSV RT-qPCR assay for the identification of PRRSV after an outbreak of the disease in swine herds, and to a lesser extent to conduct surveillance of the virus in herds. Four clinical samples were confirmed to be PRRSV positive by another veterinary diagnostic laboratory (Biovet, St-Hyacinthe, Québec, Canada). The clinical samples originated from different swine herds and types of production systems throughout the province of Quebec, Canada.
We included 149 PRRSV RT-qPCR–positive clinical samples: 80 sera (of which 48 samples are pooled sera), 33 lung tissues, 2 oral fluids (OF), and 1 processing fluid (PF; i.e., in our case, fluid from testes after castration; Table 1). An additional category of clinical samples was obtained from pathologists conducting carcass macroscopic examinations and submitted as a pool of tissues (PoT), including mainly lungs with several other types of tissues such as lymph nodes, spleen, liver, and intestine (Table 1). We included 33 PRRSV RT-qPCR–positive PoT clinical samples. The PRRSV RT-qPCR assay cycle quantification (Cq) values of all clinical samples were 11.5–34.3 (mean: 22.6 ± 4.5). More specifically, the mean Cq values of specific types of clinical samples were: 22.4 ± 3.9, 22.3 ± 4.5, 22.5 ± 4.6, 30.8 ± 2.8, and 28.9 (only 1 sample) for lung, PoT, sera, OF, and PF, respectively.
Porcine reproductive and respiratory syndrome virus (PRRSV) reverse-transcription quantitative PCR–positive sample description.
PRRSV RT-qPCR assay
The PRRSV RT-qPCR assay conducted by the MDL (American Association of Veterinary Laboratory Diagnosticians [AAVLD] accredited) was an in-house assay (protocol PON-MOL-029). Briefly, PRRSV RNA was isolated from clinical samples (QIAamp cador pathogen mini kit, Qiagen; KingFisher Flex automated nucleic acid extraction apparatus, Thermo Fisher Scientific) as recommended by the manufacturers. Thereafter, 5 μL of RNA was used to conduct RT-qPCR assays (Rotor-Gene real-time PCR; Qiagen). The primers and probe sequences of the PRRSV RT-qPCR assay cannot be disclosed because they are the property of MDL. Noteworthy, a log-linear equation to quantify the amount of PRRSV genome copies per mL or gram of clinical samples was established using a standard curve determined with commercial standards (Tetracore). The equation was established as follow: y = 10(−0.307x + 10.390) × 100; y = number of viral genome copies, x = RT-qPCR Cq value, and 100 = RNA elution volume in μL.
Genome extraction and purification
We ground 100 mg of lung or tissues (Beadbeater; BioSpec Products) in phosphate-buffered saline, then centrifuged at full speed for 1 min and used the supernatant for viral extraction. Two hundred µL of sera, OF, and PF were centrifuged for 5 min at 10,000 × g, and the supernatant was used for viral extraction. Viral RNA was extracted (Quick-RNA viral kit; Zymo Research) as described in the company’s protocol. Thereafter, RNA was eluted using 50 µL of nuclease-free water (Corning). Total elution volume was used to isolate RNA with poly(A)-tails (NEBNext poly(A) mRNA magnetic isolation module; New England BioLabs), as described in the company’s protocol, and poly(A)-tails RNA was resuspended in 15 µL of Tris buffer (New England BioLabs). Then, first-strand complementary DNA (cDNA) was synthesized (Non-directional reaction step up protocol, NEBNext RNA first strand synthesis module; New England BioLabs), starting with 10 µL of isolated poly(A)-tails RNA. Immediately after first-strand cDNA synthesis, the second DNA strand was synthesized (NEBNext Ultra II non-directional RNA second strand synthesis module; New England BioLabs) as described by the manufacturer’s protocol with a minor modification at the incubation step in the thermocycler for 2 h (instead of 1.5 h) at 16°C. Double-stranded DNA (dsDNA) was then purified (AxyPrep Mag PCR clean-up kits; Axygen Corning) using 1.8× of beads and 70% ethanol. The purified dsDNA was diluted in 30 µL of 10 mM Tris-HCl at pH 8.0 and was stored at −20°C.
PRRSV WGS
Double-stranded DNA was quantified (Qubit dsDNA HS assay kit, Qubit fluorometer; Thermo Fisher Scientific). Sequencing libraries (Nextera XT DNA library preparation kit; Illumina) were still performed even when the dsDNA quantification results were lower than the Qubit dsDNA HS assay kit threshold of detection (0.2 ng). Briefly, 0.2–0.3 ng/µL of dsDNA was used to construct the sequencing libraries (5 µL total volume was used, even when dsDNA quantification results were below the threshold of detection). Fragmentation and tagmentation were performed as suggested by the manufacturer’s protocol. Amplification and indexation were also performed as described by the manufacturer’s protocol, except that 14 amplification cycles were run. Sequencing libraries were then purified (AxyPrep Mag PCR clean-up kits) as described in the Nextera XT protocol. Library quality was assessed (High sensitivity DNA kit, Bioanalyzer; Agilent). Sequencing libraries were normalized (LNB1 beads; Nextera XT protocol), or with the manual normalization protocol if the concentrations of the libraries were in the lower part of the Bioanalyzer curves. Sequencing libraries were sequenced in a v3 600-cycle cartridge using a MiSeq instrument, and PhiX was included at ~ 1% of the total sequencing libraries as a control to establish the sequencing run efficacy (Illumina). Variable numbers of PRRSV sequencing libraries were indexed per MiSeq run in consideration of the type of samples being sequenced. In fact, several types of samples obtained from different animal species were processed simultaneously by the high-throughput sequencing laboratory and were subsequently combined, to allow optimal use of the v3 600-cycle cartridges. Those samples may have included: (1) bacteria, (2) viruses contained within various clinical samples (or may have been previously isolated), and (3) PCR amplicons (for different purposes such as 16S microbiome characterization and swine influenza A virus sequencing). Large amounts of MiSeq high-throughput sequencing reads data were obtained from each successful PRRSV WGS case (Suppl. Table 1).
Highly qualified personnel for PRRSV WGS efficiency
During our study, 2 highly qualified personnel (HQP) constructed sequencing libraries from the PRRSV RT-qPCR–positive swine clinical samples. A total of 83 and 66 clinical samples were processed by HQP-1 and HQP-2, respectively. HQP-1 processed 19 lungs, 17 PoT, and 44 sera; HQP-2 processed 14 lungs, 16 PoT, and 36 sera. HQP-1 also processed the 2 OF and the only PF clinical sample. All clinical samples that were processed by both HQPs are distinct, meaning that a specific clinical sample was processed by only one HQP, with rare exceptions. Both HQPs followed the same PRRSV WGS protocol described above.
Bioinformatic analyses
At first, reads were trimmed for adaptors and quality by the MiSeq software during FastQ generation. Using CLC Genomic Workbench software (v.12.0.3; Qiagen), reads from each sample were mapped using the Map Reads to Reference application with default settings against a list of PRRSV full-length genomes obtained from GenBank and from the PRRSV nt sequences obtained by our laboratory. Thereafter, all reads from each sample were mapped against the closest PRRSV full-length genome in the list. Consensus sequences were extracted from alignments that had full-length coverage. Moreover, reads were trimmed again for quality and adaptors using the CLC Genomic Workbench software (usually only a few reads, but some needed more trimming). This second step of trimming is highly recommended by Qiagen before doing de novo analysis. Then, de novo analysis was performed using the application “De novo Assemble Metagenome” with 2,000 minimum contigs length with the scaffolding settings option. Contigs were used to confirm the PRRSV full-length genome obtained by re-sequencing.
Reads dispersion
A typical MiSeq sequencing experiment involves fragmentation of the genome to be sequenced from a clinical sample into millions of molecules. The set of fragments, after different modifications, is referred to as a sequencing library, which is sequenced to produce a set of reads (a read length depends on the protocol and may vary 100–300 bp; BREDA Genetics. Sequencing library: what is it? 2016. Accessed 2019 Dec 17: https://bredagenetics.com/sequencing-library-cosa-e/). To calculate the reads dispersion throughout the entire viral genome (a critical step that is needed to ensure the efficiency of PRRSV WGS), the viral genome was divided into 2 sections: (1) the 5′-region of the viral genome containing the ORF1 gene and that represents ~ 75% of the entire viral genome; and (2) the 3′-region of the viral genome containing all other genes (ORF2–7). The number of reads targeting each genomic region was determined, including the overlapping reads that were included in both genomic regions. The ratio of reads was calculated as follow: 5′ viral genome-specific reads/3′ viral genome-specific reads. The higher the ratio of reads value, the higher the reads dispersion is expected throughout the viral genome.
Amount of PRRSV-specific reads
The % of PRRSV-specific reads per clinical sample was calculated as follows: (number of PRRSV-specific reads/total reads) × 100%.
Statistical analyses
All statistical analyses were performed using Prism software (v.8.3.0; GraphPad). Different types of analyses were performed, including parametric 2-tailed unpaired t tests, ordinary 1-way ANOVA with Tukey post-tests, and ordinary 2-way ANOVA.
Results
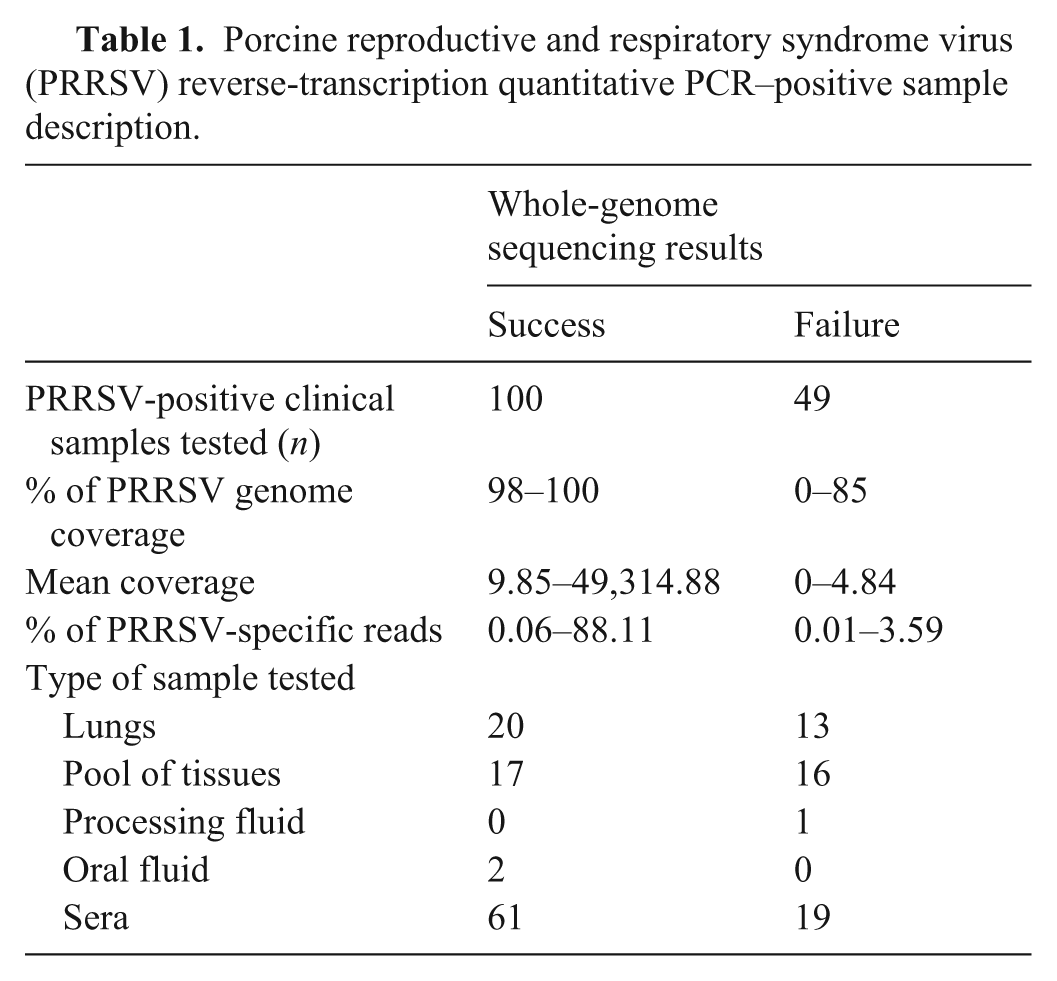
Complete PRRSV genome sequences (Table 1) were obtained from 100 samples (67.1%; Fig. 1). More interestingly, the WGS success rate was more efficient with sera (76.3%) compared to lungs (60.6%) and PoT (51.5%) clinical samples (Fig. 1). Noteworthy, OF and PF samples were not included in the statistical analyses because of the low number of samples (i.e., 2 OF and 1 PF). Nonetheless, WGS sequences of PRRSV were obtained from both OF samples, whereas the PRRSV entire viral genome was not obtained from PF.
Number of porcine reproductive and respiratory syndrome virus (PRRSV) whole-genome sequencing (WGS)-positive cases per type of clinical sample. PRRSV WGS was attempted only on PRRSV reverse-transcription quantitative PCR (RT-qPCR)-positive cases. The “all samples” group includes all types of samples that we tested (i.e., lungs, sera, OF, PF, and PoT). The number in boxes is the % of PRRSV WGS successful cases. OF = oral fluid; PF = processing fluid (i.e., castration liquid); PoT = pool of tissues; WGS (+) = PRRSV WGS success; WGS (−) = PRRSV WGS failure.
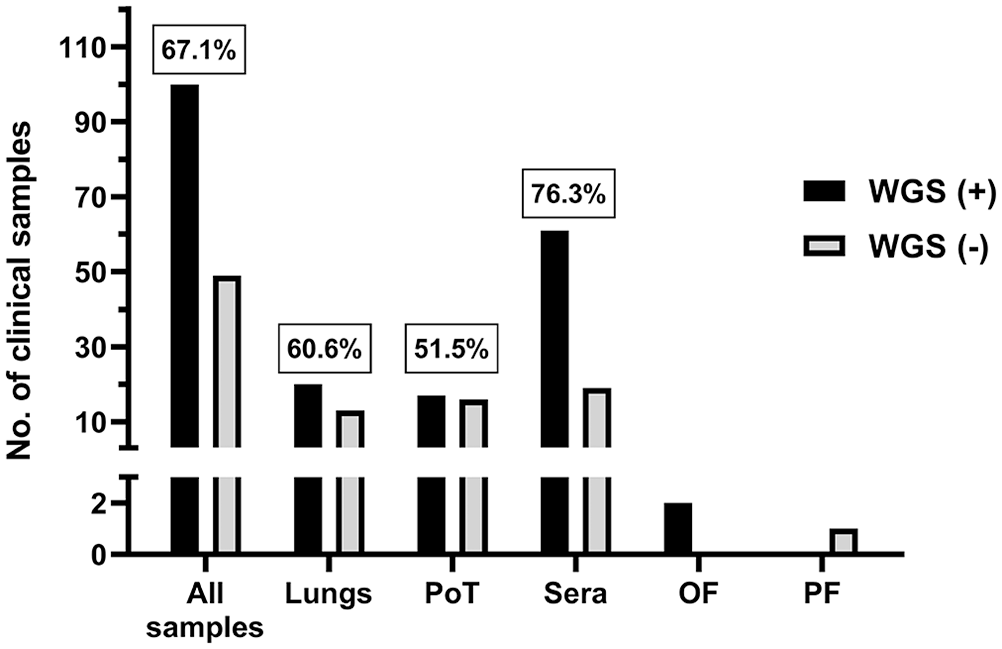
One hypothesis was that the WGS success rate is highly dependent on the viral load. To test this hypothesis and to establish the efficiency of the poly(A)-tail method for PRRSV WGS, it was essential to compare the WGS success rate with PRRSV viral load in each clinical sample. The success of PRRSV WGS from clinical samples was significantly higher with a higher viral load (i.e., lower PRRSV RT-qPCR Cq values; Fig. 2) for all types of samples (i.e., lungs, PoT and sera; p < 0.0001; 2-way ANOVA). Therefore, as expected, 100% PRRSV WGS-positive results were obtained for all types of clinical samples at the higher viral load (i.e., ≤ 15 Cq, which is the lower PRRSV RT-qPCR Cq value).
Percentage of PRRSV WGS success compared to sample viral load. The success of PRRSV WGS from clinical samples was significantly higher with a higher viral load. The “all samples” group includes all types of samples that we tested (i.e., lungs, sera, OF, PF, and PoT). Cq = cycle quantification. See Figure 1 legend for other abbreviations.
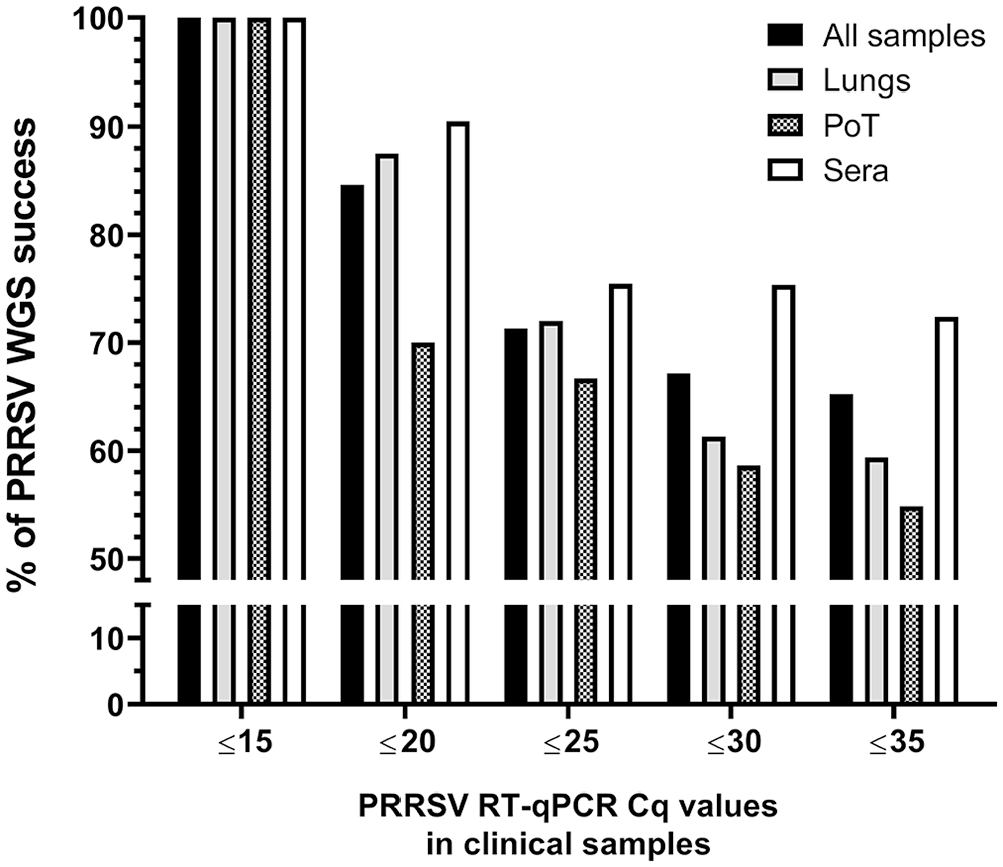
Moreover, the WGS success rate was also dependent on the type of samples being analyzed (p = 0.004; 2-way ANOVA). In fact, PRRSV WGS was more successful from sera, with and 75.4 and 72.4% WGS-positive results, for Cq values of ≤ 25 and ≤ 35, respectively (Fig. 2). The type of clinical samples from which WGS was the least successful was PoT with 66.7 and 54.8% WGS-positive results, for Cq values of ≤ 25 and ≤ 35, respectively (Fig. 2). The overall WGS success % (i.e., at Cq ≤ 35) for all clinical samples with known Cq values was 65.2% (Fig. 2). The viral load of each clinical sample was plotted and separated into 2 groups (i.e., the samples from which WGS-positive and -negative results were obtained; Fig. 3). WGS-positive results were obtained with tissues (i.e., lungs and PoT) for up to the lower viral load of 26.5 Cq, whereas for sera, the lower viral load with a WGS-positive result was 34.1 Cq (Fig. 3).
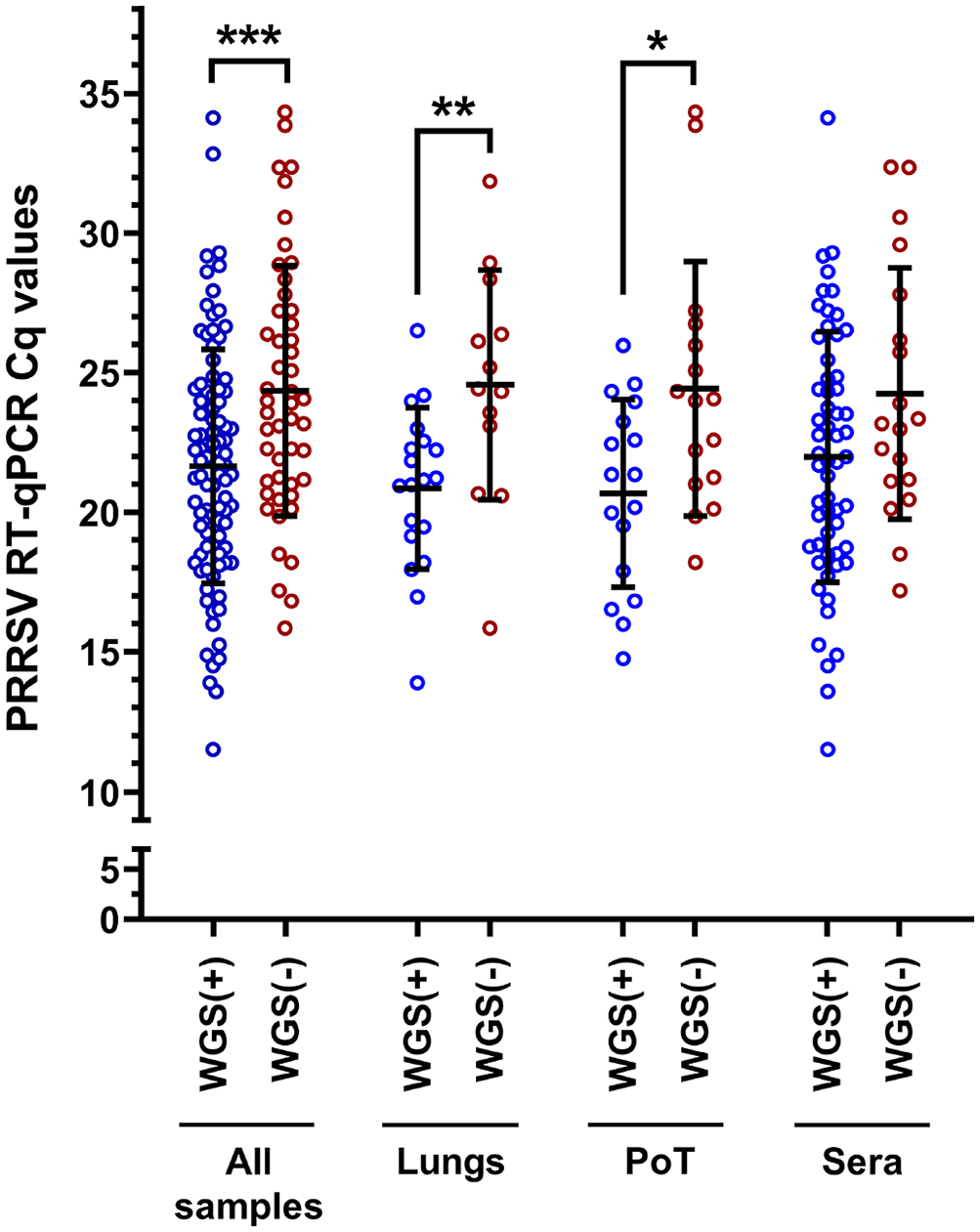
PRRSV WGS success compared to sample viral load. The “all samples” group includes all types of samples that we tested (i.e., lungs, sera, OF, PF, and PoT). The bars and whiskers represent the PRRSV RT-qPCR Cq mean ± standard deviation; the empty dots represent individual Cq sample values. WGS (+) = PRRSV WGS success; WGS (−) = PRRSV WGS failure. WGS (+) and WGS (−) groups were compared by parametric 2-tailed unpaired t tests. * = p ≤ 0.05; ** = p ≤ 0.01; *** = p ≤ 0.001. See Figure 1 legend for abbreviations.
Interesting results were obtained with OF regarding PRRSV WGS sensitivity. Although only 2 OF samples were tested, with 28.8 and 32.8 Cq values, respectively, WGS-positive results were obtained for both samples (data not shown), whereas the higher Cq values obtained with lungs and PoT were 26.5 and 26.0, respectively. Unfortunately, the only PF clinical sample tested (with 28.9 Cq value) was WGS negative (data not shown). The viral loads of WGS-positive results were significantly different compared to WGS-negative results for 2 types of samples in addition to all of the samples (Fig. 3). More precisely, the viral loads for WGS-positive and -negative cases in all samples were 21.6 ± 4.2 and 24.4 ± 4.5 (p < 0.001); in lungs, 20.9 ± 2.9 and 24.6 ± 4.1 (p < 0.01); and, in PoT, 20.7 ± 3.4 and 24.4 ± 4.6 (p < 0.05), respectively (Fig. 3). Interestingly, the viral loads in sera were not significantly different when WGS-positive and -negative results groups were compared (Fig. 3).
The efficiency and sensitivity of WGS of a virus from a clinical sample is highly dependent on the number of target-specific reads obtained during a NGS run and on the viral load contained in the clinical sample. Therefore, we evaluated and compared the % of PRRSV-specific reads obtained from clinical samples to PRRSV viral loads contained within WGS-positive and -negative samples (Fig. 4). Overall, the % of PRRSV-specific reads was higher in sera at all viral loads compared to lungs and PoT clinical samples (Fig. 4B–D). Noteworthy, the number of PRRSV-specific reads did not correlate with the PoT sample viral loads (Fig. 4C). PoT PRRSV-specific reads were very low compared to sera and lung PRRSV-specific reads. Noteworthy, PoT sample composition may differ because the pooled samples contained various amounts of different swine tissues and subsequently, may also have contained various amounts of high-throughput sequencing inhibitors. This could explain why no correlation was found between the viral loads and PRRSV-specific reads for PoT samples.
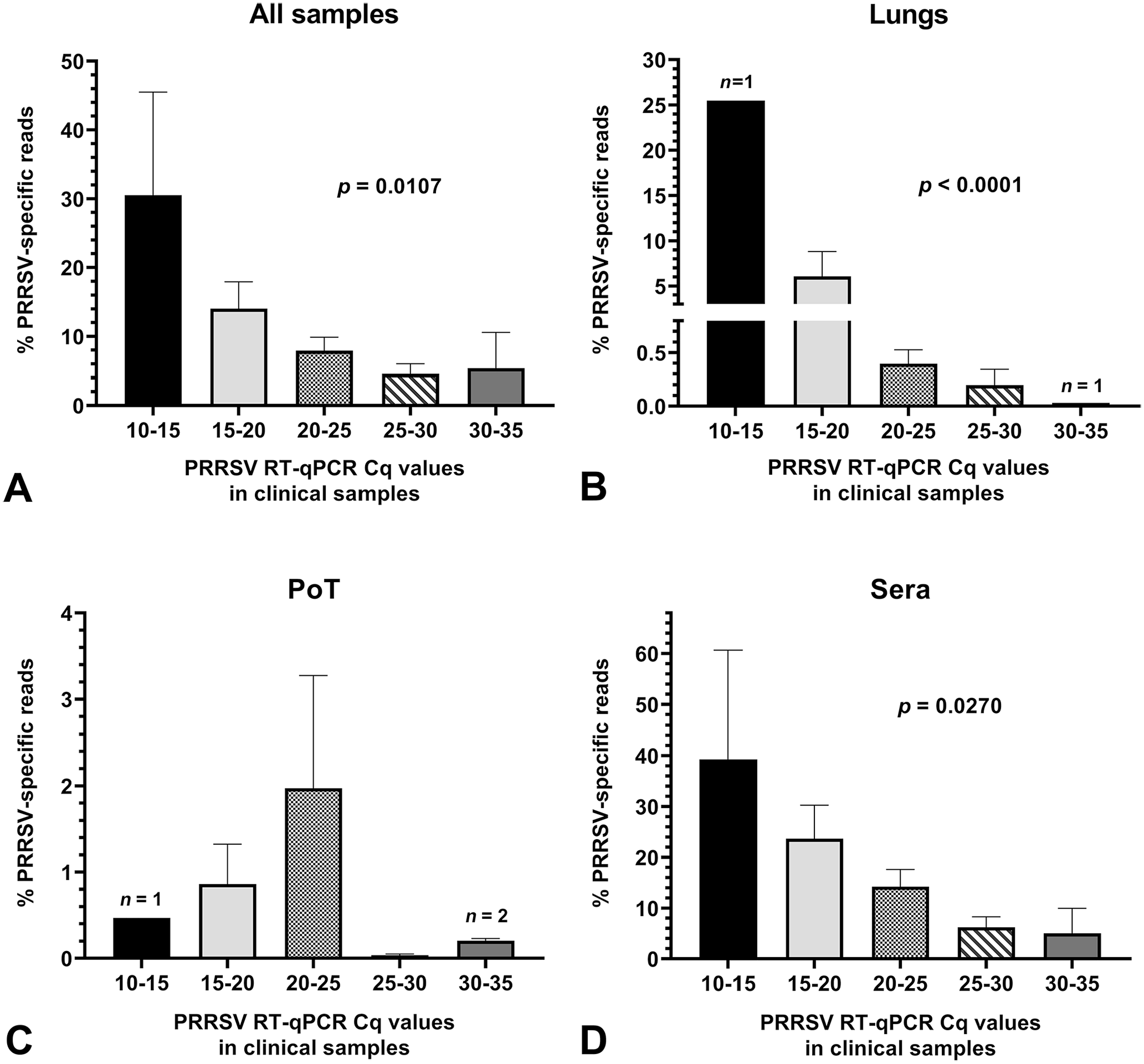
Amount of PRRSV high-throughput sequencing–specific reads in clinical samples. Graphs were built with all clinical samples (i.e. from both WGS successful and unsuccessful samples). The bars and whiskers represent the % of PRRSV-specific reads mean ± standard error of the mean. Parametric ordinary 1-way ANOVA tests were done to compare the % of PRRSV-specific reads to the clinical sample viral load.
The synthesis of sequencing libraries is a critical phase that may fail at different steps along their construction. Therefore, sequencing libraries need HQP to ensure the validity of NGS results. Consequently, we evaluated HQP efficiency (Fig. 5). Before comparing the WGS results between HQPs, it was essential to determine if the clinical sample viral loads were similar between the samples tested by each HQP. Noteworthy, the viral load of the clinical samples tested by HQP-2 was significantly higher than the viral load of clinical samples tested by HQP-1 (21.6 ± 3.9 vs. 23.4 ± 4.8, respectively; Fig. 5A; p = 0.022). Surprisingly, even if HQP-2 had tested samples with lower Cq values, their PRRSV WGS success rate was overall lower compared to HQP-1 (p = 0.021; Fig. 5B). In fact, the % of PRRSV WGS-positive results for all samples, lungs, PoT, and sera were: 78.3% and 53.0%; 79.0% and 35.7%; 58.8% and 43.8%; and 86.4% and 63.9% for HQP-1 and -2, respectively (Fig. 5B). As expected, clinical sample PRRSV Cq values were significantly lower in PRRSV WGS-positive cases compared to WGS-negative cases for both HQP (Fig. 5C).
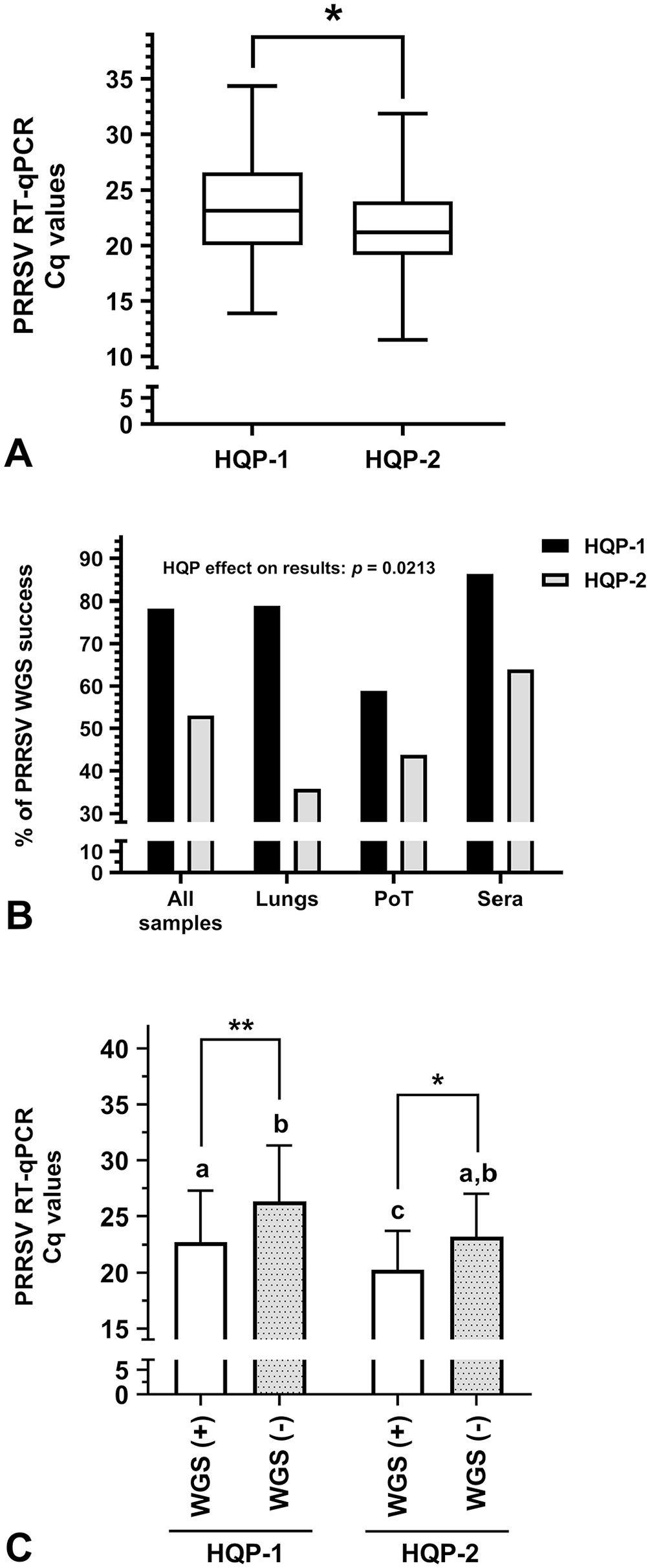
Highly qualified personnel (HQP) WGS efficiency.
In order to obtain the complete viral genome sequence of PRRSV directly from a clinical sample, it is necessary to ensure good dispersion of PRRSV-specific reads throughout the entire viral genome. Therefore, we calculated ORF1/ORF2–7 reads ratios to estimate the reads dispersion throughout the entire viral genome. Surprisingly, almost opposite results were obtained by both HQPs (Fig. 6). The reads dispersion was significantly higher in sera compared to lung clinical samples as expected for HQP-1 (p = 0.020; 1.61 ± 0.56 vs. 1.21 ± 0.54, respectively), whereas the reads dispersion seems to be higher in lungs compared to serum clinical samples for HQP-2 (1.20 ± 0.50 vs. 0.82 ± 0.87, respectively), but both sample groups were not significantly different (p = 0.359; Fig. 6). When reads dispersion results of both HQPs were combined, no statistical difference was found between serum and lung clinical samples (p = 0.459). An example of HQP-1 reads dispersion obtained with a serum and a lung sample is illustrated in Figure 6B.
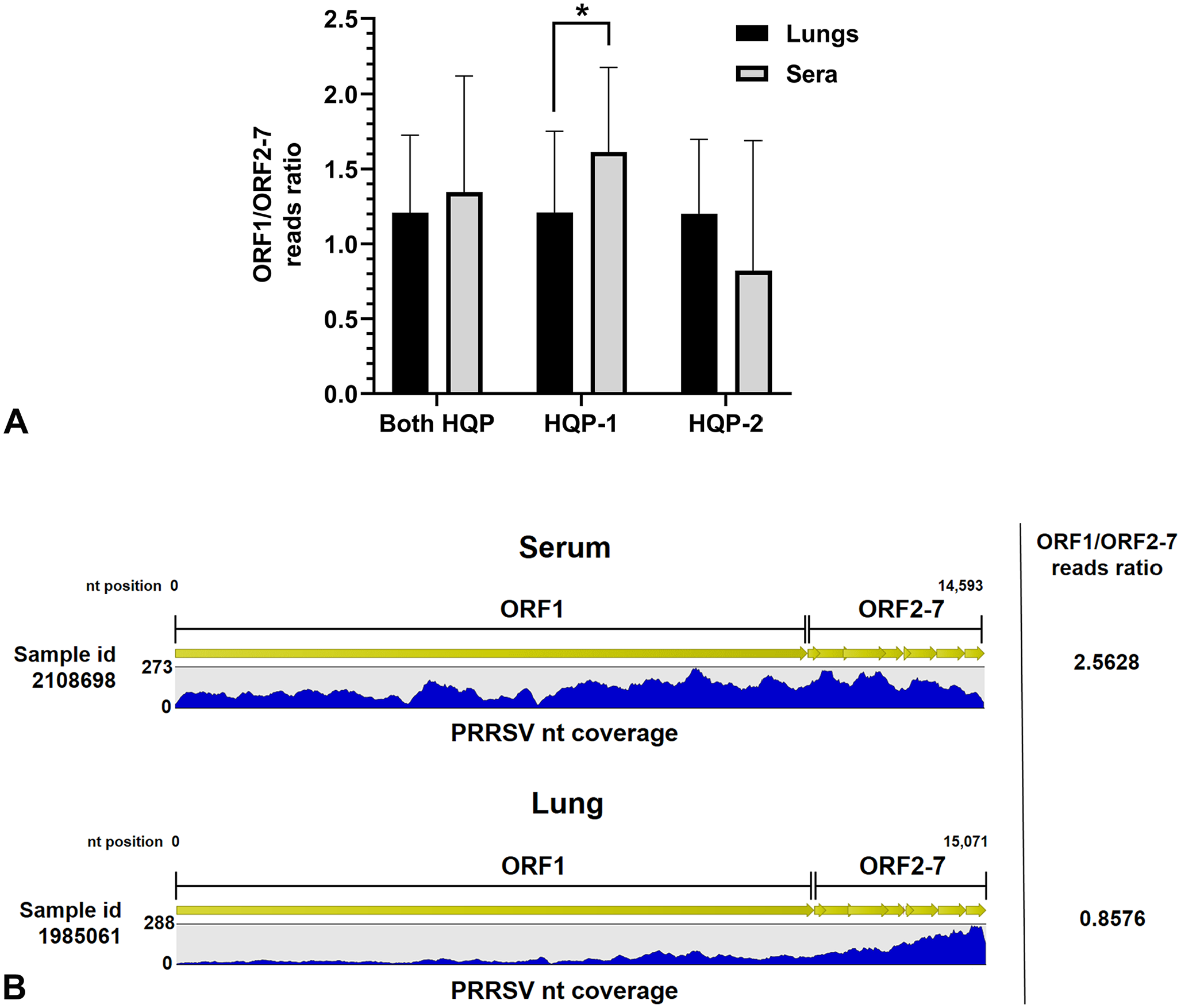
PRRSV-specific reads dispersion throughout the viral genome.
Discussion
Based on previous reports, it was obvious that NGS of the PRRSV genome from clinical samples needed to be improved significantly.17,45,48 In fact, in one research report, the metagenomic approach that was proposed to sequence PRRSV genome, which was based on Illumina technology, was unsuccessful with nasal swabs (PRRSV RT-qPCR Cq values of 25–35) and OF (PRRSV RT-qPCR Cq values of 20–29). 17 The authors suggested that these types of samples were not appropriate because of the low viral load, viral genome degradation, and competition for sequencing reads given that several other viral and bacterial genome sequences were found using their metagenomics approach, in particularly in OF. Unfortunately, in our study, no nasal swabs were included in the analyses, therefore no comparison can be made. Noteworthy, WGS of PRRSV was successful with the 2 tested OF samples even if the PRRSV viral load was lower in one sample (PRRSV RT-qPCR Cq values of 28.8 and 32.8), indicating that the poly(A)-tail approach, to increase the specific selection of PRRSV genome, has a great impact on the sensitivity of the method.
The metagenomic method that had been proposed 17 was successful for PRRSV WGS with sera in 38.6% of the tested samples (PRRSV RT-qPCR Cq values of 14–25) 17 ; whereas we achieved a success rate of 76.2%, illustrating the significant improvement that has been brought about by the poly(A)-tail method. Using a similar Illumina metagenomic method, 17 with the addition of cDNA synthesis using random primers (hexamers), others have reported the ability to generate PRRSV sequences by WGS from different types of clinical samples, but they were successful only with clinical samples containing higher viral loads compared to our study. 48 They reported PRRSV WGS to be successful only from sera with PRRSV RT-qPCR Cq ≤ 23.6 (whereas in the present study: Cq ≤ 34.1); successful only from lung tissues with Cq ≤ 21 (whereas in the present study: Cq ≤ 26.5); and successful only from OF with Cq ≤ 20.6 (whereas in the present study: Cq ≤ 32.8), 48 clearly illustrating that the poly(A)-tail method improves the sensitivity of PRRSV WGS.
In addition to MiSeq technology, others have reported the use of the Oxford Nanopore MinION direct RNA sequencing to generate PRRSV whole-genome sequences. 45 Unfortunately, in that report, only 6 swine clinical samples (all sera) were tested, reducing the statistical significance and impact of their results for field application. Moreover, the sensitivity of the Oxford Nanopore MinION PRRSV WGS with these clinical serum samples was very low 45 compared to our study. The reported sensitivity with sera of the Oxford Nanopore technology was 4.65 × 104 times less compared to the MiSeq poly(A)-tail method (Oxford Nanopore sensitivity was 3.8 × 106 viral genome copies per sequencing reaction, and PRRSV WGS was unsuccessful, compared to 8.17 × 101 viral genome copies per sequencing reaction at 34.1 Cq value with the MiSeq poly(A)-tail method, and PRRSV WGS was successful). Other investigators have also reported the low sensitivity of the direct RNA sequencing method for WGS of several RNA viruses (with or without viral poly(A) tail genomes). 46 Moreover, some concerns were raised regarding possible high sequence error rates of the direct RNA sequencing method of Oxford Nanopore technology that can limit virus strain identification. 46 The low sensitivity of the Oxford Nanopore MinION direct RNA sequencing with swine clinical samples illustrates that further investigations are needed to improve this method.
Our results demonstrate that PRRSV WGS is dependent on the viral load, a conclusion supported by other reports.17,48 Noteworthy, the viral load seems to have a negligible impact on PRRSV WGS success rate with sera (Fig. 3). Is it possible that other factors may influence the PRRSV WGS success rate with sera? Our study seems to support this hypothesis. In molecular testing, several PCR-inhibiting substances and factors may exist (such as proteases, calcium, iron, gel media, anticoagulants, degradation of the genomic content) in a clinical sample and need to be considered to ensure the quality of a result. 47 The same inhibiting factors may have an impact on WGS success from a specific clinical sample. In fact, the PRRSV WGS poly(A)-tail method was unsuccessful with some clinical samples that possess a high viral load (e.g., serum at 17.2 Cq and lung at 15.8 Cq), but overall, the number of PRRSV high-throughput sequencing reads was also higher in serum compared to lung samples at the same viral load.
It would have been interesting to determine from one individual infected pig, at different times post-infection, which is the most efficient type of sample to conduct PRRSV WGS. Unfortunately, our databank does not contain this type of sample to allow WGS efficiency comparison among samples of one infected individual. Nonetheless, we are confident that the obtained results indicate that serum is a very good sample choice to conduct PRRSV WGS.
Diverse genomes have been found by metagenomics in clinical samples (including host, bacterial, viral, and other high-throughput sequencing reads), and they are present in different ratios depending on the type of clinical sample being analyzed. 36 Authors have reported that serum is the sample type with the highest ratio of reads related to viral genome (12%, whereas 50% for host genome) compared to tissue (0.2%, whereas > 95% for host genome). 36 Therefore, it may explain why we found serum to be the most sensitive and appropriate type of sample to conduct PRRSV WGS assays, a finding also supported by 2 previous PRRSV WGS studies.17,48 It may also explain why the PRRSV WGS poly(A)-tail method, which allows the specific selection of the viral genome, may be more sensitive compared to MiSeq random primer approaches that will produce, in theory, a significantly larger quantity of indexed host genome.
The extracted RNA quality may have a major impact on PRRSV WGS efficiency. Unfortunately, the quantity of extracted and purified poly(A)-tail RNA from serum was too low to allow RNA quality assessment. In fact, it is possible to assess the RNA quality with a Bioanalyzer (Agilent) using a specific application (RNA integrity number, RIN). Unfortunately, RIN values are not accurate for samples that contain < 25 ng RNA/μL. 33 It is well known that the amount of extracted RNA from serum may be very low (i.e. 2–10 ng/μL),28,42 and therefore, we could not assess accurately the RIN value or RNA quality from serum in our study.
In theory, although PRRSV virions should be free in serum and therefore their viral genomes should not be in a replicative state, PRRSV viral genome in tissues may replicate in infected cells. During the viral genome replication of arteriviruses, a subset of viral subgenomic messenger RNA (i.e., poly(A)-tails mRNA) possessing the same 3′-end nucleotide sequences of the viral genome are synthesized. 43 Consequently, the presence of viral subgenomic mRNA in infected tissues should increase the ORF2–7 viral gene copies (these ORFs are located at the 3′-end of the viral genome) over the ORF1 viral gene copies and, therefore, reduce the ORF1/ORF2–7 read ratio. This phenomenon should subsequently decrease the high-throughput sequencing reads dispersion throughout the PRRSV viral genome from infected tissue in clinical samples. The results obtained by HQP-1 are in accordance with this theory, meaning that a better reads dispersion throughout the PRRSV genome was found with sera compared to lung samples. Unfortunately, opposite results regarding reads dispersion were obtained by HQP-2. This discrepancy may have arisen because the HQP-2 PRRSV WGS success rate was lower compared to HQP-1 even if HQP-2 tested clinical samples possessing a higher viral load. Besides the fact that PCR-inhibiting factors may be present in a clinical sample, HQP skills to conduct high-throughput sequencing laboratory handling are critical to ensure WGS assay success.
Several reports have described PRRSV WGS that were generated from viruses previously isolated in cell culture or from several PCR amplicons covering the entire viral genome.2,3,5,23 Our goal was to implement a method that can efficiently work directly from clinical samples without virus isolation or prior PCR amplicon steps because some PCR amplicons could be missing, and because mutations and isolation of more specific PRRSV cell culture–adapted strains can be promoted.11,23,31 The poly(A)-tail viral genome purification method improved significantly the efficiency of PRRSV WGS from clinical samples compared to the methods that have been reported previously.17,45,48 Furthermore, serum was the most appropriate type of sample for sequencing the entire viral genome of PRRSV.
Supplemental Material
Supplemental_material – Supplemental material for Porcine reproductive and respiratory syndrome virus whole-genome sequencing efficacy with field clinical samples using a poly(A)-tail viral genome purification method
Supplemental material, Supplemental_material for Porcine reproductive and respiratory syndrome virus whole-genome sequencing efficacy with field clinical samples using a poly(A)-tail viral genome purification method by Carl A. Gagnon, Christian Lalonde and Chantale Provost in Journal of Veterinary Diagnostic Investigation
Footnotes
Acknowledgements
We thank all of the veterinarians who have submitted clinical samples to their university diagnostic laboratory; their trust in the FMV diagnostic service is tremendously appreciated.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
This research was financially supported by the Ministère de l’Agriculture, des Pêcheries et de l’Alimentation du Québec (MAPAQ) Innov’Action program. C.A. Gagnon was financially supported by a Natural Sciences and Engineering Research Council of Canada (NSERC) discovery grant and a Canadian Swine Research and Development Cluster (CSRDC) grant. C. Lalonde was a recipient of a scholarship from the CRIPA, a research network financially supported by the Fonds de recherche du Québec–Nature et technologies (FRQNT).
Supplementary material
Supplementary material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.