
Abstract
Pattern-recognition receptors (PRRs) are expressed by innate immune cells and recognize pathogen-associated molecular patterns (PAMPs) as well as endogenous damage-associated molecular pattern (DAMP) molecules. With a large potential for synergism or convergence between their signaling pathways, PRRs orchestrate a complex interplay of cellular mediators and transcription factors, and thus play a central role in homeostasis and host defense. Aberrant activation of PRR signaling, mutations of the receptors and/or their downstream signaling molecules, and/or DAMP/PAMP complex–mediated receptor signaling can potentially lead to chronic auto-inflammatory diseases or development of cancer. PRR signaling pathways appear to also present an interesting new avenue for the modulation of inflammatory responses and to serve as potential novel therapeutic targets. Evidence for a dysregulation of the PRR toll-like receptor (TLR)2, TLR4, TLR5, and TLR9, nucleotide-binding oligomerization domain–containing protein (NOD)2, and the receptor of advanced glycation end products (RAGE) exists in dogs with chronic enteropathies. We describe the TLR, NOD2, and RAGE signaling pathways and evaluate the current veterinary literature—in comparison to human medicine—to determine the role of TLRs, NOD2, and RAGE in canine chronic enteropathies.
Keywords
Introduction
The innate immune system is the first-line host defense mechanism. It consists of cells and molecules that can respond rapidly to a variety of microbes recognized by differences from “self” cells (so-called pathogen-associated molecular patterns [PAMPs]) and to endogenous host-derived danger- or damage-associated molecular patterns (DAMPs, or alarmins). Thus, innate immunity is a complex system of circulating cells, sentinel cells, circulating molecules, and cellular molecules that generate a reaction with the purpose of neutralizing invading microbes. 37
Aberrant signaling within the innate immune system, including the pattern-recognition receptor (PRR) signaling pathways, has the potential to cause chronic auto-inflammatory diseases or development of cancer. Evidence for dysregulation of the PRR toll-like receptor (TLR)2, TLR4, TLR5, and TLR9, nucleotide-binding oligomerization domain–containing protein (NOD)2, and the receptor of advanced glycation end products (RAGE) exists in dogs with chronic enteropathies (Kathrani A, et al. Overdominant single nucleotide polymorphisms in the nucleotide oligomerisation domain two (NOD2) gene are significantly associated with canine inflammatory bowel disease [abstract]. J Vet Intern Med 2010;24:725; Kathrani A, et al. Mutational analysis of the TLR5 gene in Boxer dogs with inflammatory reveals novel polymorphisms in the leucine rich repeat domain [abstract]. J Vet Intern Med 2011;25:696).1,6,11,13-15,17-20,26,31,43 We describe the TLRs, NOD2, and RAGE signaling pathways and evaluate the current veterinary literature—in comparison to human medicine—to determine the role of TLRs, NOD2, and RAGE in canine chronic enteropathies.
Pattern-recognition receptors
Several different PRRs, such as TLRs, NOD2, and RAGE, are expressed by innate immune cells and recognize PAMPs and DAMPs. With a large potential for synergism or convergence between their signaling pathways, PRRs orchestrate a complex interplay of cellular mediators and transcription factors, and thus appear to play a central role in homeostasis and host defense.
Structure of TLRs, NOD2, and RAGE
Some TLRs (TLR1, TLR2, TLR4, TLR5, TLR6, and TLR10) and full-length RAGE are cell surface (transmembrane) proteins with a similar yet distinct structure (Fig. 1). Other TLRs (TLR3, TLR7, TLR8, TLR9, TLR11, TLR12, and TLR13) and NOD2 are intracellular (cytoplasmic) PRRs with some (NOD2, TLR9, some chimeric TLRs)3,4 also localizing to the cell membrane. Transmembrane TLRs consist of 1) an ectodomain of leucine-rich repeats (LRRs) that mediates PAMP and/or DAMP recognition and has a horseshoe-like structure, 2) a transmembrane domain functioning as an anchor, and 3) the cytoplasmic domain toll/interleukin-1 (IL-1) receptor (TIR), which initiates downstream signaling (Fig. 1). 4 The NOD2 receptor, an intracellular receptor, has 4 domains: 1) a LRR-domain that determines protein-to-protein interactions, 2) a nucleotide-binding oligomerization domain (NOD), and 3–4) two caspase-recruitment domains (CARDs) responsible for initiating downstream pathways. 31 Full-length RAGE consists of 5 domains: 1) a variable (V-type) domain responsible for ligand binding, 2–3) two constant (C-type) domains, 4) a transmembrane domain (anchor), and 5) a cytoplasmic domain that initiates signal transduction (Fig. 1). 35
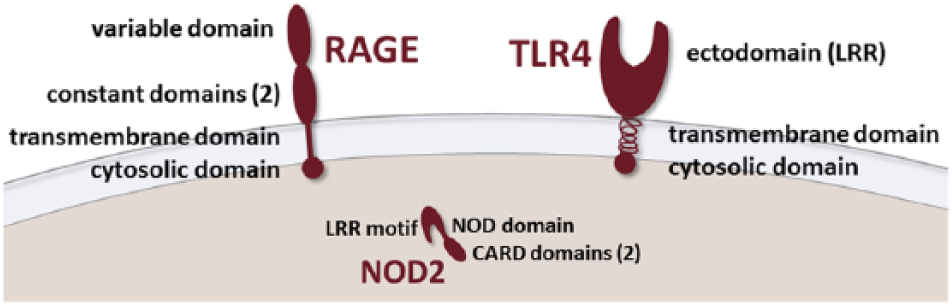
Structure of the receptor for advanced glycation end products (RAGE), the transmembrane toll-like receptor 4 (TLR4), and the cytoplasmic nucleotide-binding oligomerization domain–containing protein (NOD)2. CARD = caspase activation and recruitment domain; LRR = leucine-rich repeats.
Natural alternative splicing or protease cleavage (e.g., during tissue damage or inflammation 16 ) can produce isoforms of cell surface receptors that lack the cytosolic domain (e.g., dominant-negative RAGE), isoforms lacking a ligand-binding domain (e.g., N-truncated RAGE), or decoy receptors (e.g., soluble RAGE [sRAGE]) that can sequester ligands and prevent their interaction with cell surface (transmembrane) receptors. The presence of these truncated receptor variants can modulate or abrogate cell signaling, and thus abolish the effect of receptor ligands.5,12,16
Ligands of TLRs, NOD2, and RAGE
Most TLRs predominantly recognize distinct PAMPs. As an example, TLR4 recognizes predominantly bacterial lipopolysaccharide [LPS], TLR5 recognizes bacterial flagellin, and TLR9 recognizes unmethylated 2’-deoxyribo-cytidine-phosphateguanosine [CpG] DNA motifs. However, there is no absolute specificity of ligands for individual TLRs, and some overlap exists between the different TLR ligands, which is also determined by the type and localization of the receptor. 4 The ligand for NOD2 is the muramyl dipeptide (MDP) molecule, a peptidoglycan component of gram-positive as well as gram-negative bacteria, and potentially also viral constituents such as single-stranded RNA.22,33 In contrast to most TLRs and NOD2, RAGE can bind several different ligands, such as advanced glycation end products (AGEs, a heterogeneous group of non-enzymatically altered proteins that accumulate at sites of inflammation), S100/calgranulin-like molecules (calcium-binding proteins of the EF-hand superfamily), high-mobility group box 1 (HMGB-1, or amphoterin, a DNA-binding protein and cytokine mediator), amyloid β protein (which plays a role in Alzheimer disease), Mac-1 (CD11b/CD18, also known as integrin or complement receptor-3), and phosphatidylserine. Multi-ligand RAGE has also been shown to bind bacterial LPS, and, for some ligands, the presence of glycans and/or a redox state appears to be essential for mediating the ligand–RAGE interaction. 42
Several TLR/RAGE ligands (e.g., HMGB-1 and LPS) can also form high molecular weight complexes with the ability to elicit stronger responses than the individual participating partner molecules alone. Binding affinity and preferential receptor activation by some TLR/RAGE ligands (e.g., S100 proteins) also depend on the cell type, the concentration and 3D-conformation of the ligand, and the pathophysiological state of the organism and/or tissue.
Signaling pathways of TLRs, NOD2, and RAGE
RAGE, NOD2, and TLR/myeloid differentiation factor-88 (MyD88) signaling pathways play a role in the immune response. Several different intracellular signal transduction pathways exist, coupling the extracellular signals presented by the different receptor ligands into different cellular responses. Post-receptor signaling pathways of TLRs, NOD2, and RAGE involve the activation and nuclear translocation of nuclear factor–kappa B (NF-κB), leading to the downstream activation of innate immune responses as an immediate host defense response (e.g., against microbes), modulation of adaptive immune responses (i.e., against specific antigens), and cell growth and proliferation or apoptosis.28,39
Depending on the receptor type and its ligand(s), TLRs recognize PAMPs and/or DAMPs as a receptor homo- or heterodimer with co-receptors or accessory molecules. Ligand recognition by TLRs leads to the recruitment of an adaptor protein, either MyD88 or TIR domain-containing adaptor–inducing interferon-β (TRIF).
MyD88-dependent signaling pathway
MyD88 recruits the protein kinases IRAK-4 and IRAK-1 (also referred to as myddosome), which associate with the adaptor protein TRAF6. The IRAK1–TRAF6 complex dissociates from the TLR and activates mitogen-activated protein kinases (MAPK) and I-κB kinase (IKK), the latter of which leads to the activation and nuclear translocation of NF-κB. In addition, MyD88 interacts with the transcription factor interferon regulatory factor (IRF)-5 and activates it by phosphorylation (Fig. 2).36,42
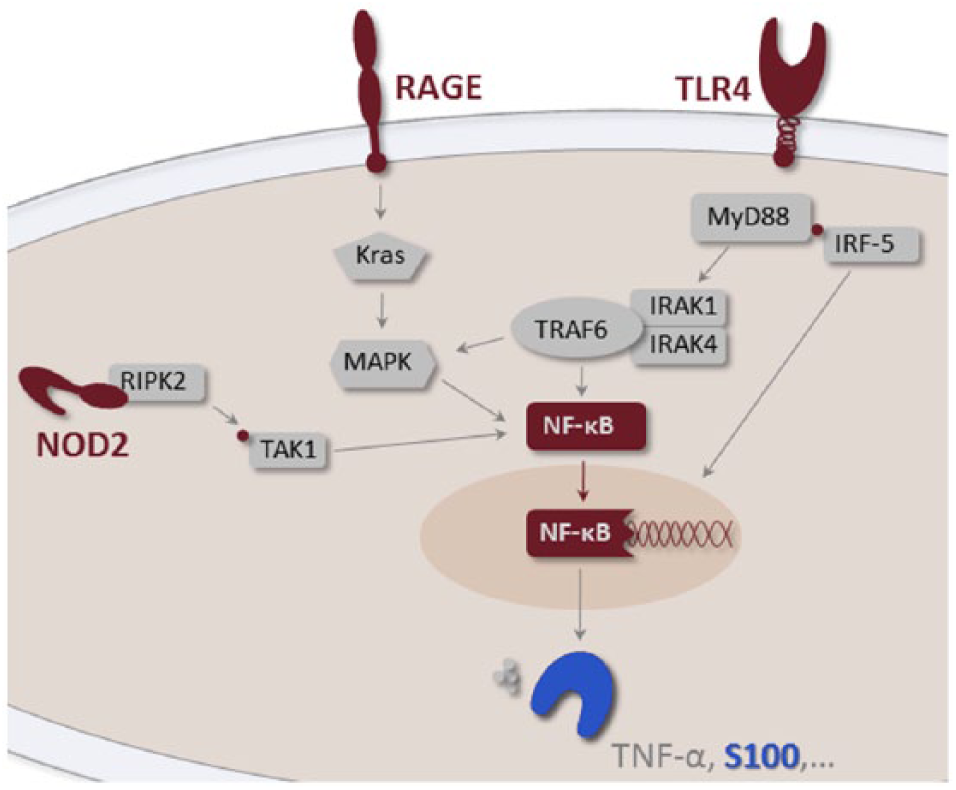
RAGE, NOD2, and TLR/MyD88 signaling pathways. Both RAGE and TLR recognize PAMP and DAMP molecules. Upon binding of those ligands, both innate immune receptors activate NF-κB, leading to the production of cytokines (pro-inflammatory and/or interferon-β) and chemokines. A positive feedback on RAGE expression has also been shown. 5 NOD2 is an intracellular PRR, which, following self-oligomerization, associates with RIPK2 and can also induce the NF-κB signaling pathway or induce the autophagy pathway. IRAK = IL-1 receptor–associated kinase; IRF = interferon regulatory factor; Kras = V-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog; MAPK = mitogen-activated protein kinases; MyD88 = myeloid differentiation factor-88; NF-κB = nuclear factor–kappa B; NOD2 = nucleotide-binding oligomerization domain–containing protein 2; RAGE = receptor for advanced glycation end products; RIPK2 = RICK protein kinase; TLR4 = toll-like receptor 4; TNF-α = tumor necrosis factor–α; TRAF = TNF receptor–associated factor.
TRIF-dependent signaling pathway
TRIF associates with TRAF3 and recruits the kinases IKK-ε and TBK-1, both of which activate transcription factor IRF-3 via phosphorylation. TRIF can also directly associate with and activate TRAF6, resulting in the activation of MAPK and IKK, but this is to a lesser extent than via the MyD88-dependent signaling pathway. 21
Activation of TLRs culminates in the activation and nuclear translocation of NF-κB as well as the activation of IRFs and MAPKs (e.g., p38 or JNK). 36 Most TLRs, with the exception of TLR3 (which recognizes double-stranded RNA), signal through MyD88 and induce the expression of proinflammatory cytokines (e.g., TNF-α, IL-1). TLR3 signals through TRIF and induces interferon-β. 21 TLR4 is a dual receptor and signals through both pathways, but instead of associating directly with MyD88 or TRIF, it uses 2 additional adaptor proteins (TIRAP which binds to MyD88, and TRAM which associates with TRIF). 21
TLR signaling is controlled by negative regulators targeting specific molecules of the TLR signaling pathways, and TLR trafficking after de novo synthesis in the endoplasmic reticulum is regulated by several mechanisms. 24 These autoregulatory feedback loops prevent or terminate excessive immune responses that could be detrimental to the host and lead to auto-inflammation, maintaining a fine balance between positive and negative regulation of TLR signaling.
NOD2 transduces signals by interaction of its CARD domains with the CARD domain of the RICK protein kinase (RIPK2) leading to the phosphorylation of transforming growth factor β–activated kinase 1 (TAK1), followed by activation of MAPK and IKK and thus activation and nuclear translocation of NF-κB (Fig. 2). 22 The LRR domain of NOD2, as well as RICK, also appears to have a regulatory role through TAK1, likely preventing overt NF-κB activation. 9 In addition, binding of NOD2 to the ATG16L1 protein and their co-localization at the plasma membrane induces the autophagy signaling pathway. 38 Autophagy is a regulated self-degradation of intracellular components by lysosomal enzymes. However, mediators of inflammation can modify (i.e., induce or inhibit) autophagy signaling pathways. 7
Similar to TLRs, signaling pathways of RAGE also lead to the activation of several kinases (MAPK p38/Erk, and NF-κB), resulting in the activation and nuclear translocation of NF-κB.5,41 Several signaling pathways, including the KRAS signaling pathway (Fig. 2), have been identified to require RAGE. 41 However, RAGE can also signal through the MyD88-dependent pathway through interaction with TIRAP and MyD88. 41
Role of TLRs, NOD2, and RAGE in host defense
The PRR TLRs, NOD2, and RAGE can signal individually and independently from each other, but they may also functionally interact. In light of several shared ligands (e.g., HMGB-1, LPS, or S100 proteins), the potential synergism and convergence between TLR and RAGE (and likely also NOD2) signaling pathways may modulate the downstream activation of transcription factors and potentially amplify associated cellular responses, thus altering the pathophysiologic state of the tissue.8,39
NOD2 plays an important role in autophagy, which is essential for the degradation of intracellular proteins as well as ingested bacteria, and leads to the formation of an autophagosome around the invading microbe, and thus in antigen processing and presentation. TLR2 and TLR4 are also capable of initiating autophagic mechanisms, and augmentation of TLR responses by NOD2 signaling can result in synergistic stimulatory post-receptor and inflammasome activity (inflammasomes are multimolecular complexes incorporating proteins that orchestrate enzyme activation and cytokine release and are involved in host defense and innate and adaptive immune responses 32 ). In addition, NOD2 responses can present a second line of host defense in the face of TLR tolerance. However, NOD2 can also inhibit and thus regulate innate immune responses, as reported in a previous review. 8
RAGE is constitutively expressed in the lung and skin, whereas in other cell types (e.g., macrophages or endothelial cells) RAGE is induced by either accumulation of its ligands and/or activation of transcription factors (e.g., NF-κB) that regulate RAGE expression. 5 Thus, the expression of RAGE coincides with a proinflammatory microenvironment, and the upregulation of this PRR increases the recruitment of inflammatory cells through an increased expression of endothelial adhesion molecules (e.g., VCAM-1, ICAM-1). 5 The decoy receptor sRAGE (a truncated isoform of the transmembrane receptor produced by natural alternative splicing or protease cleavage [Heilmann RM. Evaluation of canine S100A12 and sRAGE as novel disease markers in dogs with inflammatory bowel disease. PhD thesis. Texas A&M University, 2015]) can sequester RAGE ligands such as DAMPs, thus preventing their interaction with cell surface RAGE and also with other PRRs. 5
TLRs, NOD2, and RAGE signaling pathways orchestrate a complex interplay of cellular mediators and transcription factors, which is important for homeostasis and host defense. PAMP signaling is crucial for host defense responses to infection, whereas the response to cell components released in response to inflammation and tissue injury (DAMPs) is an essential mechanism to stimulate cytokine production and activate cells of the innate immune system (particularly macrophages and dendritic cells) in order to alert the immune system of imminent danger. However, aberrant activation of TLR, NOD2, and/or RAGE signaling, mutations of the receptors and/or downstream signaling molecules (i.e., positive and/or negative regulators), and/or DAMP/PAMP complex–mediated TLR signaling can potentially lead to a loss of controlled homeostatic tolerance and sustained cellular inflammatory responses, causing chronic auto-inflammatory or neoplastic conditions. 39 TLR, NOD2, and RAGE signaling pathways appear to present an interesting avenue for the modulation of inflammatory responses (e.g., via PRR agonists, antagonists, neutralizing antibodies, or gene silencing tools) and serve as potential novel therapeutic targets.
Role of TLRs, NOD2, and RAGE in inflammatory bowel disease in humans
Evidence of dysregulated PRR signaling has been demonstrated in people with inflammatory bowel diseases (IBDs; i.e., Crohn’s disease and ulcerative colitis). We provide a brief summary of the comparatively relevant PRR dysregulations detected in patients with IBD.
Polymorphisms in the TLR4, TLR5, and TLR9 genes have been associated with Crohn’s disease, whereas TLR2 single nucleotide polymorphisms (SNPs) are associated with ulcerative colitis, as reported in a previous review. 39 Expression of TLR2 and TLR4 has been shown to be increased in the intestines of people with active IBD, suspected of rendering a pro-inflammatory state in the intestinal immune system. 39
NOD2 mutations can result in either a loss- or a gain-of-function and have been associated with several inflammatory disorders, including IBD.8,22,39 Similar to the LRR SNPs in TLRs, polymorphisms affecting the LRR domain of the NOD2/CARD15 gene have been associated with an increased susceptibility for Crohn’s disease. These polymorphisms may result in an impaired recognition of muramyl dipeptide (MDP) necessary for the control of innate immune responses. 8 These polymorphisms can also cause a transition of NOD2 into its constitutively activated state, impaired regulation of autophagic or cross-tolerogenic TLR responses, and/or reduced production of intestinal α-defensin, cryptin, and/or IL-10 as a result of NOD2 dysfunction. 8 NOD2 mutations are currently considered one of the strongest risk factors for the development of Crohn’s disease in people. 22
Also, polymorphisms in the NOD2-adaptor protein ATG16L1 have been associated with Crohn’s disease likely associated with a dysregulation of the autophagy pathway. 8 A SNP in the locus of NLRP3 (another cytoplasmic NOD-like receptor that does not represent a true PRR but is a component of a cytoplasmic multiunit complex (inflammasome) involved in the initiation of apoptosis and transcription of several pro-inflammatory cytokines) has also been associated with an increased risk of developing Crohn’s disease in people. 39
Studies in people with auto-inflammatory conditions, including IBD, have revealed a perpetuation and amplification of the inflammatory response caused by ligand–RAGE interaction and sustained RAGE signaling 5 ; decreased systemic sRAGE levels, especially in patients with ulcerative colitis (whether this reflects a decrease of constitutive levels and/or is dynamic with inflammation remains to be determined)10,27; and an association between RAGE polymorphisms and the susceptibility to ulcerative colitis. 40 Taken together, these findings support the current concept that the pathogenesis of IBD in people involves the complex interaction of a number of dysregulated PRR signaling pathways.
Role of TLRs, NOD2, and RAGE in canine chronic enteropathies
Research to evaluate defects and dysregulations in the innate immune receptors (PRRs) as potential contributors to the pathogenesis of canine chronic enteropathies (CE) has made great progress in the past decade. Several gene expression studies and the evaluation of candidate genes revealed that some PRR dysregulations are comparable with those found in people with Crohn’s disease or ulcerative colitis.
Activation of NF-κB, a key regulator of the transcription of several inflammatory and immunoregulatory genes, has been demonstrated in dogs that were diagnosed with CE.25,30,31 No correlation was seen between NF-κB and the severity of clinical signs25,30,31 or histologic lesions30,31 in these CE dogs. The response to treatment was associated with a downregulation of NF-κB activation. 25 Dogs that responded to an elimination diet alone (food-responsive disease) showed higher pretreatment NF-κB activation within the duodenal epithelium compared to dogs that required anti-inflammatory and/or immunosuppressive therapy (IBD). 25
Increased duodenal and colonic mucosal expression of TLR2, TLR4, and TLR9 messenger (m)RNA and down-regulation of TLR5 expression have been reported in dogs with IBD where there was no correlation with either histologic severity of inflammation or response to treatment.1,6,26 Discrepant results were obtained for an increased TLR4 gene expression in dogs with CE,6,26 which could be attributed to the difference in sample size or reference genes used and the fact that increased expression of TLR mRNA may not be simply related to the number and/or type of inflammatory cells within the intestinal mucosa of affected dogs.6,26 Conflicting results for the relationship between TLR expression and clinical disease activity were also obtained,1,6,26 where only TLR2 expression levels showed a weak linear correlation with the severity of clinical signs in one study. 26 However, in none of these 3 studies were the findings confirmed at the protein level, and the cell type within the mucosa expressing increased TLR mRNA levels was not determined.1,6,26
Inflammatory colorectal polyps, proposed to be a novel breed-specific form of canine IBD that, to date, has only been reported in Japanese Miniature Dachshunds, were also associated with an increased mucosal expression of TLR2 and TLR4 mRNA, 13 both of which were localized to inflammatory cells and colonic epithelium within the affected mucosa. 43 Further, stimulation of TLR2 was enhanced in peripheral blood-derived monocytes from affected Dachshunds, 15 demonstrating a functional relationship.
Polymorphisms in the genes encoding TLR4, TLR5, and NOD2 were also significantly associated with CE in German Shepherd dogs and other breeds, as reported in a previous review. 2 Two TLR4 SNPs (A1571T and G1807A, both recessive) and a TLR5 SNP (G22A; additive, possibly dominant) have been associated with an increased risk of IBD in German Shepherds, whereas 2 TLR5 SNPs (C100T, dominant in German Shepherds and other breeds; T1844C, dominant in German Shepherds but determined to be recessive in a study including 38 different dog breeds) were protective for IBD independent of the breed.17,18 An additional SNP (T443C) in the LRR domain of TLR5 was associated with IBD in Boxer dogs (Kathrani A, et al. Mutational analysis of the TLR5 gene in Boxer dogs) suggesting unique polymorphisms in individual breeds in addition to shared SNPs among dog breeds. Significance of the TLR5 SNPs G22A (risk-associated) and C100T/T1844C (risk-protective) in dogs with IBD revealed a hyper-responsiveness of TLR5 to stimulation with flagellin (gain-of-function and loss-of-function, respectively) as assessed using a NF-κB luciferase assay and CXCL8 ELISA. 22 Carrying these risk-associated or risk-protective SNPs was not associated with the number of immunoglobulin A (IgA)-positive cells in the duodenum or colon of German Shepherds with CE, 23 but secretory IgA levels were not evaluated in the previous study. Taken together, these findings support a role of TLR5 in canine CE and agree with the results of studies in human IBD and experimental models. Evaluation of TLR5 as a potential therapeutic target in dogs with CE warrants further research.
Mucosal NOD2 mRNA levels were increased in dogs with lymphoplasmacytic colitis in one breed-independent study. 31 In contrast, an upregulation of mucosal NOD2 mRNA levels could not be demonstrated in duodenal mucosal samples from dogs (several breeds) with lymphoplasmacytic enteritis. 30 NOD2 is the only PRR that has been evaluated in relation to NF-κB activation in dogs with CE.30,31 The lack of correlation between the latter and NOD2 mRNA in the intestinal mucosa supports a complex regulation of NF-κB activation.30,31 Levels of NOD2 mRNA were also not correlated with the severity of clinical signs or histologic lesions in either study.30,31 Expression of mucosal NOD2 mRNA and NOD2 reactivity in peripheral blood-derived monocytes were also increased in Miniature Dachshunds with inflammatory colorectal polyps.13,15
Heterozygosity for 4 NOD2 SNPs (G1357A, C1578T, G1693C, A1885G) located in exon 4 were found to be associated with IBD in dogs (Kathrani A, et al. Overdominant single nucleotide polymorphisms in the nucleotide oligomerisation domain two (NOD2) gene). These findings suggest a significant role of NOD2 in canine IBD. Mutational analyses in German Shepherds also showed a breed-dependent association of IBD with 5 polymorphisms in the LRR domain of the NOD2 gene (A1537G, T1578C, C1639G, and G1885A in exon 3; and G2569A in exon 6) that was not found in IBD dogs of other breeds. 20 However, the implication of those 5 NOD2 SNPs on NOD2 function requires further investigation. Four non-synonymous SNPs (A1532G, T1573C, C1688G, G1880A; all protective) were also detected in NOD2 exon 3 in Miniature Dachshunds with inflammatory colorectal polyps, 14 and these SNPs may play a role in the development of this disease.
In contrast to a mouse model of experimental colitis, 39 expression of the protective NOD-like receptor NLRP3 was found to be decreased in dogs with CE. 34 Although the functional significance of this finding for intestinal homeostasis and epithelial integrity remains to be further investigated, the increased IL-1β protein expression in the intestinal lamina propria and its decrease after treatment in dogs with food-responsive diarrhea seen in the study 34 provides evidence for an activated inflammasome pathway in canine CE.
Canine RAGE and its naturally occurring splicing variants (total of 24) have been characterized, and the expression of canine RAGE in different tissues has been described.29,35 Similar to findings in people with IBD, systemic sRAGE concentrations were also decreased in dogs with IBD. 11 Serum sRAGE concentrations did not correlate with the severity of clinical signs or histologic lesions, nor with the response to treatment or patient outcome. 11 An interesting result in that study was that serum sRAGE concentrations only increased in IBD dogs that experienced a complete clinical remission during the course of treatment. 11 These findings suggest a potential dysregulation in the sRAGE/RAGE axis in dogs with IBD. However, to our knowledge, the expression of RAGE at the tissue level (possible polymorphisms in the RAGE gene) and the sRAGE/RAGE axis have not been studied to date in canine CE, and this requires further research.
Conclusion
PRR signaling pathways are important for host defense and homeostasis; dysregulation in this complex network may lead to sustained inflammatory responses. Previous studies have shown dysregulation of several PRRs in the intestines of dogs with CE, suggesting that innate immunity is in a state of hyperreactivity, which, similar to the findings in human IBD, may contribute to the pathogenesis of chronic inflammation in dogs with CE. Whether some of these PRR dysregulations are a cause or consequence of chronic gastrointestinal inflammation remains to be determined. Future research is also necessary to determine the contribution of PRRs that have not yet been investigated extensively (e.g., the sRAGE/RAGE axis) and to evaluate the complex interplay between these PRR dysregulations and their role in the pathogenesis of canine CE. Further research is also warranted to determine whether specific decoy receptors (e.g., sRAGE) offer a potential therapeutic target.
Footnotes
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.