
Abstract
Dogs with a 4-bp deletion in the MDR1 (or ABCB1) gene show intolerance to certain drugs routinely used in veterinary medicine, such as ivermectin, vincristine, and doxorubicin. The mutation leads to a dysfunctional P-glycoprotein drug transporter, which results in drug accumulation in the brain and severe neurotoxicity. A rapid and accurate in-house test to determine the genotype of patients in cases of acute neurotoxic signs or in tumor patients is desirable. We describe a cost-effective detection method with simple technical equipment for veterinary practice. Two allele-specific methods are presented, which allow discrimination of all genotypes, require little hands-on time, and show the results within ~1 h after DNA sampling. DNA from buccal swabs of 115 dogs with known genotype (no mutation, n = 54; heterozygous for the mutation, n = 37; homozygous for the mutation, n = 24) was extracted either by using a column-based extraction kit or by heating swabs in a simple NaOH-Tris buffer. Amplification was performed either by allele-specific fast polymerase chain reaction or by allele-specific loop-mediated isothermal amplification (LAMP). Analysis was done either on agarose gels, by simple endpoint visualization using ultraviolet light, or by measuring the increase of fluorescence and time to threshold crossing. Commercial master mixes reduced the preparation time and minimized sources of error in both methods. Both methods allowed the discrimination of all 3 genotypes, and the results of the new methods matched the results of the previous genotyping. The presented methods could be used for fast individual MDR1/ABCB1 genotyping with less equipment than existing methods.
Introduction
A 4–base pair (bp) deletion mutation in the gene for the drug transporter P-glycoprotein (formerly multidrug resistance gene [MDR1], now referred to as ATP-binding cassette subfamily B member 1 [ABCB1] gene) is found with a very high allele frequency in several sheepdog breeds, with >70% in the Collie breed and ~50% in Australian Shepherd dogs.13,22 Other breeds of Collie descent and mixed-breed dogs with Collie ancestry are affected to a lesser extent. 7 In our clinic, purebred dogs of these 3 breeds and mixed dogs with sheepdog and collie ancestry represent 2.5% of canine patients.
Because the gene product of the deleted allele has been found to be dysfunctional, treatment with drugs that are substrates for P-glycoprotein causes severe or even fatal neurotoxic reactions in individuals homozygous for the deletion. 12 Some of these drugs, such as ivermectin, vincristine, and doxorubicin, are frequently used in veterinary medicine. A fast and accurate genotyping method for the ABCB1 gene status of individual dogs could be used to preempt cases of acute neurotoxicity and in choosing safe therapeutic options. Several polymerase chain reaction (PCR)–based methods for allelic discrimination are used to date, but have not reached a format that may be used as a point-of-care detection test.1,6,11,16 Ideally, an in-house test for allele-specific DNA amplification would be based on noninvasive DNA sampling and require minimal hands-on time and limited technical equipment for DNA extraction, amplification, and detection.
In 2000, an isothermal amplification method (loop-mediated isothermal amplification [LAMP]) was developed. 19 Four primers, matching 6 specific sequences, form products with self-hybridizing loop structures. The reaction does not require melting or annealing steps, given that the DNA polymerases used display strand displacement activity. At a constant amplification temperature of 60–65°C, DNA fragments of different lengths are formed, which can be detected by various methods. The technique was improved by including additional stem primers or loop primers to facilitate and accelerate the reaction.5,18 The method has been found to be quite robust and less affected than PCR by inhibitors found in crude sample material. 10 Because of its high sensitivity, technicians should avoid opening the reaction tubes after amplification to decrease the risk of carryover contamination. 23 Because LAMP reactions require only simple heat blocks, and DNA amplification can be detected by fluorescence or color change, the method can be applied for point-of-care testing.3,20 Although numerous LAMP reactions for the detection of infectious agents have been described, only a few studies have been published using this method for the detection of DNA mutations.2,4,9,15
Although reliable amplification methods for MDR1/ABCB1 genotyping do exist, a simple and cost-effective test for individual patients could improve diagnosis in cases of suspected intoxication and help select adequate drugs in tumor patients. In our study, we developed 2 rapid methods for allele-specific MDR1/ABCB1 genotyping that were compared against the results of official genotyping tests. The results of both methods, allele-specific PCR with fast amplification and allele-specific LAMP with fluorescent detection of amplicons, matched the expected genotypes of every individual. From sample to result, both methods could be completed in ~60 min.
Materials and methods
DNA sampling and extraction
For DNA sampling, buccal epithelial cells were collected using sterile dry cotton swabs a from 115 dogs with known genotypes (no mutation, MDR1 (+/+), n = 54; heterozygous for the mutation, MDR1 (+/–), n = 37; homozygous for the mutation, MDR1 (–/–), n = 24; Table 1). All dogs had been tested for their genotype by a licensed commercial laboratory (TransMIT GmbH, Gießen, Germany) or were offspring from tested parents with a homozygous genotype (both parents MDR1 (+/+) or MDR1 (–/–)). The test is patent-protected, and details are not disclosed. Two swabs per dog were rotated against the inside of the cheek, dried for 10 min, sealed, and stored at −20°C. Permission to use these samples was granted by the ethics commission of the Centre for Clinical Veterinary Medicine at the Ludwig Maximilian University Munich (5-05-29-13).
Breed distribution of collected samples.
DNA from 98 swabs was extracted using a commercial extraction kit b and eluted with 150 µL of buffer AE (10 mM Tris-Cl and 0.5 mM EDTA; pH 9.0). To test the feasibility of a simple and inexpensive extraction protocol, 40 swabs were extracted using a variation of a protocol created for noninvasive genotyping of mice. 14 The swabs were soaked with 28 µL of 0.1 M NaOH, clipped off, and incubated for 10 min at 75°C in a reaction tube containing 252 µL of Tris buffer (20 mM Tris-HCl pH 8.0). The swabs were removed, and the supernatant was used as a template without further purification. Extracted DNA samples were stored at −20°C until further processing.
DNA amplification
For allele-specific (AS)-PCR, the total reaction volume of 20 µL included 10 µL of PCR master mix, c 5 µL of template, 4 µL of water, 0.5 µL of allele-specific forward primer (F_wtMDR or F_mutMDR), and 0.5 µL of reverse primer (R_MDR) at a concentration of 25 pmol/µL each (Table 2). The forward primer was specific either to the wild-type or to the mutant allele, and each sample was tested in 2 separate reactions. The amplification was performed on a thermocycler with fast ramping rates d with the following cycling protocol: hot start at 95°C for 5 min; 45 cycles of denaturation at 98°C for 5 s, annealing at 50°C for 5 s, synthesis at 68°C for 10 s, and extension at 72°C for 1 min. The reaction was completed in 35 min and generated amplicons of 335 or 331 bp, respectively. All 115 dogs with known genotypes were tested (Table 1).
Primers used for allele-specific (AS)-PCR and AS-LAMP.*

Lowercase letters in sequences represent the base mismatches. LAMP = loop-mediated isothermal amplification; PCR = polymerase chain reaction.
The primer design for AS-LAMP was assisted by the software PrimerExplorer. e Two allele-specific reactions were performed for each sample. The reaction mix contained 15 µL of master mix, f 5 µL of primer mix, 3 µL of water, and 2 µL of template for a total volume of 25 µL. The primer mix consisted of 1 µM F3, 1 µM B3, 4 µM FIP_wt or FIP_mut, 4 µM BIP, 2 µM stemB, and 2 µM stemF (Table 2). The reaction mix was incubated at 61°C for 75 min in a real-time PCR system. g The total reaction time of 75 min was chosen to detect unspecific amplification. Sixty-five samples, which had all been extracted using the extraction kit, were tested (25 MDR1 (+/+), 21 MDR1 (+/–), 19 MDR1 (–/–)).
Analysis
PCR products were visualized on 2% agarose gel. h Alternatively, 1 µL of a 1:100 GelRed i dilution was added to the AS-PCR tubes after amplification, and the tubes were examined under ultraviolet (UV) light j at 312 nm. During AS-LAMP, accumulation of DNA products was measured by detecting the increase of fluorescence in the FAM detection channel (λmax 518 nm), and the time to threshold crossing was analyzed. A subset of samples was analyzed on 2% agarose gel to demonstrate the formation of DNA fragments by the LAMP reaction.
Results
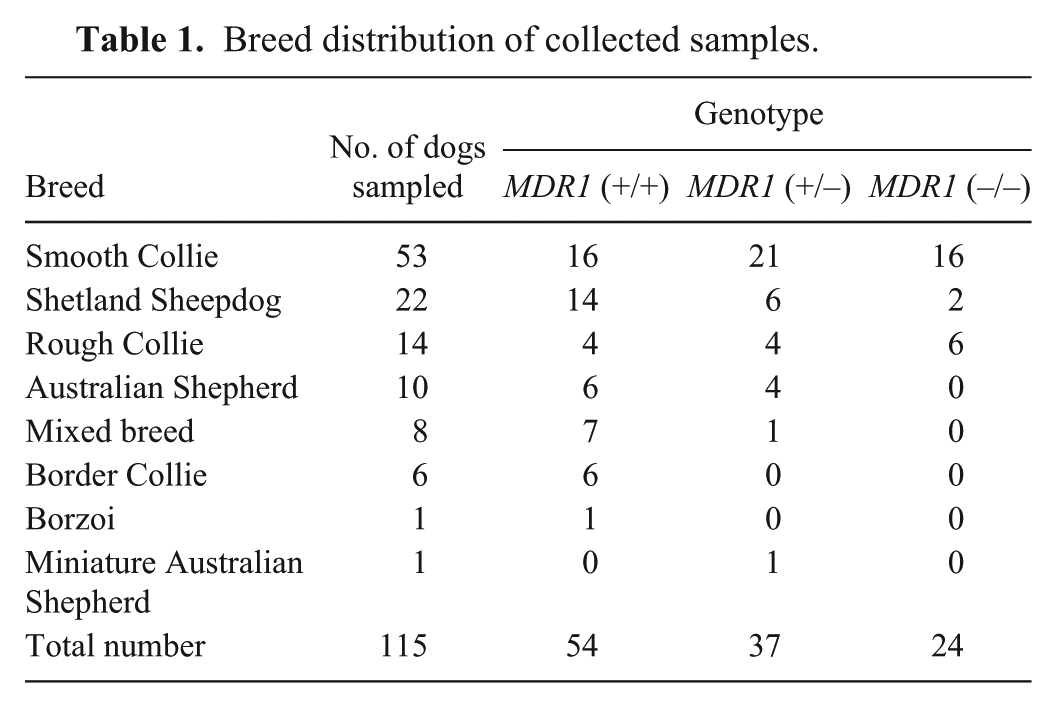
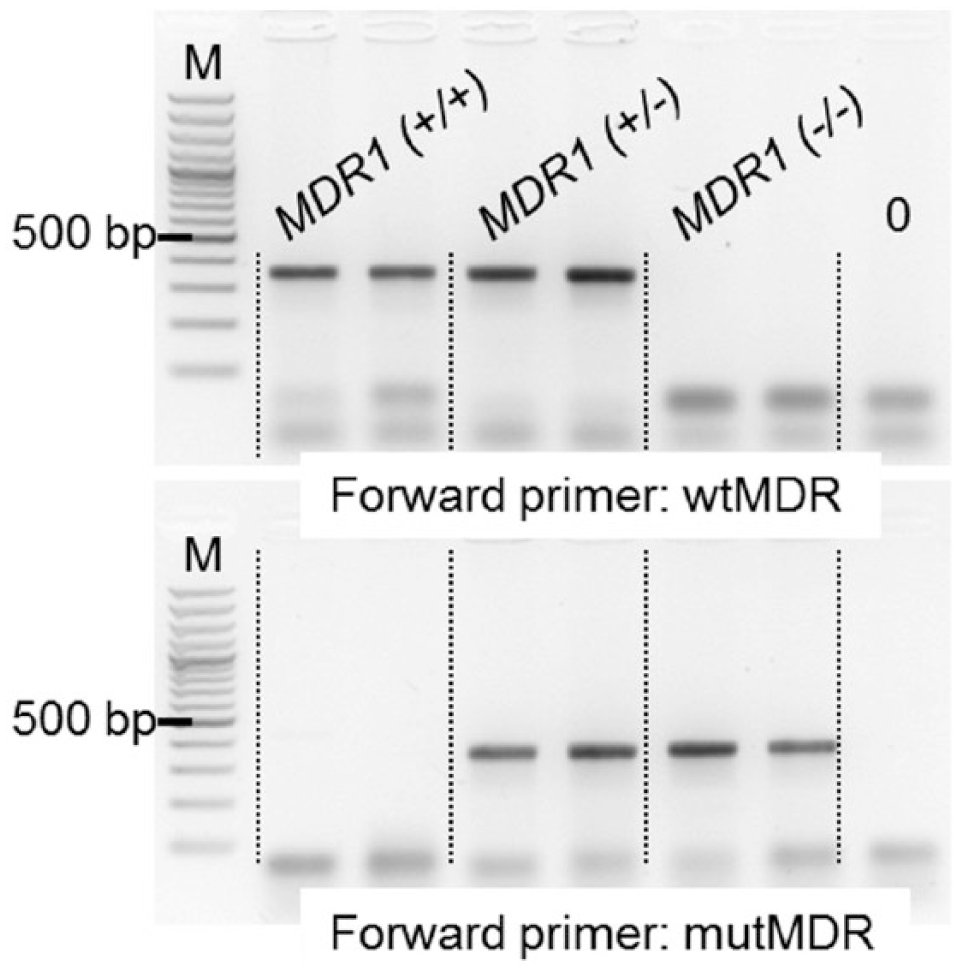
The fast allele-specific PCR yielded the expected results for all individuals when analyzed on an agarose gel (Fig. 1). The amplification products of DNA samples from swabs extracted with a commercial kit could also be visualized directly under UV light by adding GelRed to the reaction tube after amplification (Fig. 2). DNA from swabs that were only heated in buffer was sufficient as a template when the reaction product was visualized on a gel, but did not yield enough DNA for direct visualization with GelRed.
Allele-specific (AS)–polymerase chain reaction products from MDR1 (+/+), MDR1 (+/–), and MDR1 (–/–) dogs on 2% agarose gel. M: 100-bp marker ladder. Upper panel: amplification with a primer specific for the wild-type allele; lower panel: amplification with a primer specific for the mutant allele.
Allele-specific (AS)–polymerase chain reaction products from MDR1 (+/+), MDR1 (+/–), and MDR1 (–/–) dogs using GelRed and UV light for visualization. Upper panel: amplification with a primer specific for the wild-type allele; lower panel: amplification with a primer specific for the mutant allele.
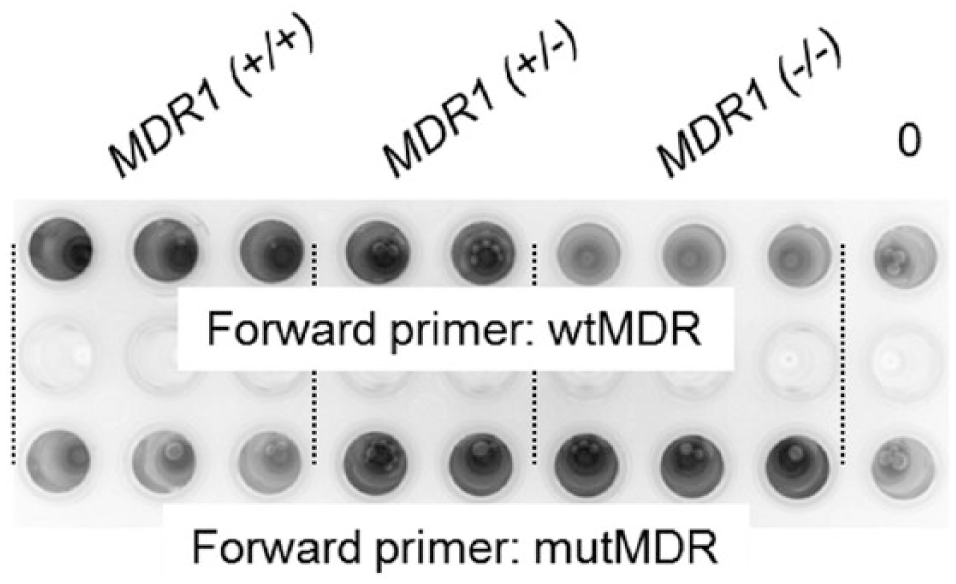
The LAMP reaction could also be used for genotyping when the times to threshold crossing between the reactions containing the FIP primer for the wild-type and the primer for the mutation for each individual were compared. The amplification efficiency of the wild-type primer was higher than that of the primer for the mutation. Time to threshold crossing was dependent on DNA content. The wild-type allele in MDR1 (+/+) and MDR1 (+/–) animals was detected in 26–43 min. The mutant allele in MDR1 (+/–) and MDR1 (–/–) animals was detected in 34–50 min. Ten of 25 MDR1 (+/+) and 1 of 19 MDR1 (–/–) samples did not show any unspecific amplification after 75 min. In 15 MDR1 (+/+) samples, an unspecific signal was detected with the primer for the mutation after 50–69 min, which was at least 18 min later than the corresponding specific signal. In 18 MDR1 (–/–) samples, a nonspecific signal with the wild-type primer was detected after 43–70 min, but always (at least 3 min) later than the specific signal. In heterozygous samples, the wild-type allele was always detected before the mutated allele, with a minimal time between detection of both alleles of 1 min and a maximum time of 11 min. Using these cutoffs, all samples could be assigned to the correct genotype (specificity 100%).
Fixed cutoffs of time to threshold without comparison between both amplifications of 45 min for the wild-type allele and 55 min for the mutant allele would have misclassified 2 MDR1 (+/+) samples and 1 MDR1 (–/–) sample as heterozygous (specificity 95% and 97%, respectively). Figure 3 shows LAMP amplification reactions that were terminated after 55 min and run on an agarose gel. The shorter run time could suppress nonspecific amplification.
Allele-specific (AS)–loop-mediated isothermal amplification products from MDR1 (+/+), MDR1 (+/–), and MDR1 (–/–) dogs on 2% agarose gel. M: 100-bp marker ladder. Upper panel: amplification with a primer specific for the wild-type allele; lower panel: amplification with a primer specific for the mutant allele.
Discussion
Major limitations for PCR-based methods as point-of-care tests are the rather time-consuming DNA extraction, long amplification protocols, and the need for specialized equipment for amplification and detection of PCR products. In our study, we tried to overcome some of these limitations to assess the MDR1/ABCB1 genotype of dogs accurately and quickly with methods that might be used in a clinical setting.
Noninvasive DNA sampling by buccal swabs is a fast and simple method that can be carried out by dog owners or clinic staff. To facilitate the extraction step, instead of a column-based extraction, some buccal swabs were heated in a simple buffer, which yielded enough DNA for AS-PCR. A specialized PCR machine with high ramping rates was used to shorten amplification. Detection using an agarose gel always yielded a reliable result, independent of the extraction method. However, this detection method needs equipment and hands-on time hardly compatible with a point-of-care test. After amplification, DNA could also be detected by adding an intercalating dye and using a UV lamp, which is a simple and fast method. However, reliable differentiation between positive and negative reactions was only possible when DNA concentrations were high enough, which needed higher sample DNA input that could not be achieved by the simple extraction in heated buffer. More sensitive and simple DNA detection is needed for samples with limited DNA content. Advances in label-free detection of DNA molecules with more sensitive dyes might overcome this drawback, or primers labeled with tag-spacer and biotin might be used for low-cost and simple detection with single tag hybridization on chromatographic printed array strips. 17
The AS-LAMP method could also be used for fast genotyping. Because opening LAMP reaction tubes after amplification is not advisable, direct detection methods built into the amplification device are preferred. Several small point-of-care amplification and detection devices are available. The isothermal master mix used in our study is designed for a portable device with heat blocks delivering a constant temperature, as well as fluorescent detection at 510 nm. k The real-time PCR system used in our study was programmed to hold a constant temperature for a comparable reaction setup. Although nonspecific amplification did occur, the differences in time to threshold between the reactions for the wild-type and the mutant allele were large enough to distinguish the 3 genotypes. Several samples were analyzed 2 or 3 times in different reaction setups with changes in time and temperature, and yielded reproducible results. Amplification detection in LAMP reactions could be further simplified if the reaction could be adapted to run in a setup with pH-sensitive dyes, leading to equipment-free visual detection. 21
Different primer sets were tested for AS-LAMP, which showed variable specificity and sensitivity. The higher number of primers needed for LAMP makes primer design more challenging compared to PCR. The PrimerExplorer software helps the investigator choose the best combinations, but adequate binding sites may be limited. In our study, the best results were achieved with FIP primers including mismatched bases close to the 5′-end in the region of the mutation site. In the recommendations for LAMP primer design, selecting this region should improve the specificity of the primers to distinguish between wild-type and mutant sequences. In the AS-PCR reactions, forward primers with comparable variations close to the 3′-end were used, given that they had been shown to work well in a previous study. 16 Instead of loop primers, which are most often used in LAMP reactions, so-called stem primers were introduced as proposed in an earlier study to achieve fewer restrictions of binding site options. 5 Although sensitivity and specificity were good for the wild-type FIP primer, which produced only little nonspecific amplification for the mutant allele after long incubation times, the mutant FIP primer appeared to be less efficient and less specific. Further variations of the primer sequence might improve the specificity of the mutant FIP primer. An alternative approach could be the inclusion of a single sequence-specific fluorescent probe in the LAMP reaction and the analysis of the melting curves to distinguish wild-type and mutant alleles. 8
Footnotes
Acknowledgements
We thank the study dog owners and clinical staff for their cooperation.
Authors’ contributions
CP Stiedl contributed to acquisition, analysis, and interpretation of data. K Weber contributed to conception and design of the study; contributed to analysis and interpretation of data; and critically revised the manuscript. Both authors drafted the manuscript, gave final approval, and agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
a.
Swabs 155 × 12 mm, sterile (80.1301), Sarstedt, Nümbrecht, Germany.
b.
QIAamp DNA mini kit, Qiagen, Hilden, Germany. Protocol: DNA Purification from Buccal c. Swabs (spin protocol) in QIAamp DNA mini and blood mini handbook 11/2007.
c.
Fast Cycling PCR master mix, Qiagen, Hilden, Germany.
d.
Gradient S thermocycler, Eppendorf, Hamburg, Germany.
f.
Isothermal master mix, OptiGene, Horsham, United Kingdom.
g.
7500 real-time PCR system, Applied Biosystems, Foster City, CA.
h.
TopVision agarose (R0491), Thermo Fisher, Waltham, MA.
i.
GelRed, Biotium, Hayward, CA.
j.
UV transilluminator, Major Science, Saratoga, CA.
k.
Genie II, OptiGene, Horsham, United Kingdom.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.